Muscle–eye–brain disease
Muscle–eye–brain (MEB) disease, also known as muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A3 (MDDGA3),[2] is a kind of rare congenital muscular dystrophy (CMD), largely characterized by hypotonia at birth. Patients have muscular dystrophy, central nervous system abnormalities and ocular abnormalities. The condition is degenerative.
| Muscle–eye–brain disease | |
|---|---|
| Other names | Muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A3 [1] |
 | |
| Muscle–eye–brain disease has an autosomal recessive inheritance. | |
| Specialty | Neurology |
| Usual onset | Birth or infancy |
MEB is caused by mutations in the POMGnT1 gene, it is congenital and inherited as an autosomal recessive disorder. There is no cure for MEB. Supportive care mainly focuses on symptoms alleviation which varies in different clinical settings. Symptomatic treatment may include physiological therapies, occupational therapies, medications, and surgeries. The life expectancy of patients with MEB is over 70 years old.
Signs and symptoms
The main signs and symptoms of MEB includes:[3]
- Muscle dystrophies: muscle weakness, hypotonia, muscle atrophy
- Ocular abnormalities: lack of visual response, severe myopia, glaucoma
- Central nervous system abnormalities: intellectual disability, cortical malformation
Muscle dystrophies
The most prevalent signs of MEB is infants being born floppy.[4] It refers to the condition of hypotonia. The types of hypotonia found on patients include generalized hypotonia, diffuse hypotonia, congenital hypotonia and the other subtypes.[5] Its cause behind is mainly the severe muscular dystrophy and partly brain abnormalities.[6]
80% - 99% individuals display various myopathy.[2] In general, they manifest severe muscle weakness and retarded motor development. Hence, gait disturbance (abnormal walking) is found in 80% - 99% of the diseased individuals.[2] A significant portion of them have limited mobility that fail to walk or even rotate the head.[4] The muscle weakness affects facial muscles apart from skeletal muscles. It gives rise to speech impairment in most of the cases.
The other signs include joint and/or spinal rigidity, reduced muscle mass, hyporeflexia, muscular contracture, spasticity, muscle atrophy and spinal deformities.[4]
Ocular abnormalities
Visual problems are often found in people with MEB. Patients have low visual acuity and fail to fixate to the visual stimuli. Depends on the severity, some display no visual response, some can response to light and some to object.[7]
More than 80% of the patients reported visual impairment.[2] Common problems include glaucoma, myopia, strabismus (“crossed-eye”) and optic atrophy which all occurs in 80-99%.[2] Optic atrophy contains poor pigmentation of ocular fundi, thin blood vessels of retina, small optic discs and scleral border and optic coloboma (degeneration of the inferior optic nerve).[3][8] Cataract affects 30% - 79% of individuals.[2] Buphthalmos (enlargement of the eyeball), megalocornea (enlargement of cornea) and nystagmus (uncontrollable eyeball movement) are present in some rare case.[2][8]
Although ocular abnormalities account for the poor vision to a large extend, some of the visual problems is associated with the brain abnormalities.[7]
Central nervous system abnormalities
MEB impairs the cognition gravely. This is reflected by the abnormal EEG and EMG reading in 80% - 99% individuals.[2] At the same frequency, hydrocephalus present in many cases.[2] The severe form of it is associated with the compression of other nerves and results in more complications.[9]
Clinical features include severe mental retardation in all aspects. Most individuals have intellectual disabilities of a range of severity.[2][10] Progressive deterioration in behavioral development has been recorded.[8] Epilepsy and seizures are found in 30% - 79% of the affected individuals.[2]
Manifestations in structural malformation are common as well. Hypoplasia of mesencephalon, pons, cerebellum and medulla are often.[11][12] Aplasia may occur on top of hypoplasia. Flattened brainstem, ventriculomegaly, pachygyria, Type II lissencephaly have been reported.[10][12] At the rate between 5% and 29% holoprosencephaly and meningocele occurs in patients too.[2]
Physical appearances
Distinctive facial features for MEB found are high prominent forehead, bulging eye and narrow temporal regions.[13]
Causes
MEB is caused by mutations of the protein O-linked mannose β1,2-N-acetylglucosaminyltransferase 1 (POMGnT1) gene. Pathogenic variants mutate the gene and lead to dysfunction of the POMGnT1 enzyme. Currently, there are 14 clinically identified mutations, the locations are dispersed and scattered throughout the POMGnT1 gene.[10][6] Different mutations were observed in MEB patients from different countries, namely Finland, Sweden, Norway, Estonia, USA, Israel, Spain and Italy.[6] From 14 non-Finnish patients, 9 different types of mutation were identified.[6]
The location of the mutation is slightly correlated with the severity of the symptoms in terms of brain structural abnormalities.[10] Mutations close to the 5′ terminus of the POMGnT1 coding region lead to relatively more severe phenotype such as hydrocephalus. Mutations that occurred close to the 3′ terminus shows weaker symptoms.[10]
MEB is an autosomal recessive disease inherited from parents. Patients with MEB have two copies of a pathogenic variant in their gene. There will be a risk of having a child with MEB given that both parents are carriers of a pathogenic variant in their gene. The affected child inherits a mutated copy of the gene from each carrier parent. The chance of inheritance is 1 in 4 when both parents carry the pathogenic variant.[15]
Pathophysiology
The pathogenesis of MEB is related to an abnormal level of α‐dystroglycan glycosylation. Genetic mutations of the POMGnT1 gene reduced the O-mannosyl glycosylation of α-dystroglycan. The POMGnT1 gene encodes the enzyme POMGnT1, a type II transmembrane protein residing in the Golgi Apparatus.[14] The role of the enzyme POMGnT1 is to catalyse glycosylation specific for alpha-linked terminal mannose, the process where N-acetylglucosamine is added to O-linked mannose of α-dystroglycan.[14] In humans, O-mannosylation is a rare type of glycosylation, occurring in skeletal muscle, brain and nerve glycoproteins.[10] O-mannosylation is used to increase the stability of the interaction between the extracellular basement membrane and α-dystroglycan. Without stabilization, the glycoprotein cannot anchor to the cell, leading to congenital muscular dystrophy (CMD), characterised by severe brain malformations.[16]
Diagnosis
Medical diagnosis for the MEB usually involves the study of family history, measurement of serum CPK level, molecular testing, muscle biopsy and imaging study.[17]
Physical examination
People with MEB have distinctive facial dysmorphisms.[18] Rounded forehead, thin and drooping lip, micrognathia, midface retrusion, short nasal bridge are the possible indicative evidence for diagnosis.[2][18] Assessment of motor and mental development, visual ability also provide clues.
Genetic test
Genetic test can analyze the genome of infants for confirmation of the specific genetic mutation. Mutation in the POMGNT1 is the determinant in the diagnosis of MEB.[19] Several mutations like [c.1539+1G→A], [c.879+5G→T] are the prevalent nucleotide change found in affected people.[18][19] The commonly used practices collect fetal DNA by chronic villus sampling, followed by linkage analysis and direct sequencing to conclude the POMGNT1 gene sequence.[20]
The genome determination helps to distinguish other congenital muscular dystrophies before and after birth. However, only some laboratories provide prenatal genetic test to screen for MEB.[2]
Medical imaging
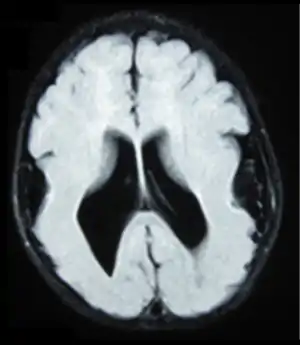
MEB can be diagnosed with medical imaging by the shared patterns of brain structural abnormalities. Common practice includes magnetic resonance imaging (MRI) and computerized tomography (CT). They can show the enlargement of ventricle, absence or degeneration of septum pellucidum, pachygyric symptoms, abnormalities in corpus collosum, lissencephaly.[21] Fetal MRI and ultrasound are used as a prenatal diagnostic tool if needed to screen for the disease. Observation like general structural malformation in the third trimester suggests congenital muscular dystrophy.[22] Further diagnostic test is required to make confirmation.
Clinically, MRI is preferred over CT scan for its ability to reveal the neuron migration more precisely.[21] The result obtained from CT scan is limited to the size of ventricles and location of white matter whereas only MRI can provide information on cortical problems.
Muscle biopsy
Muscle biopsy is a means to investigate the muscle tissue. Morphology of muscle cells and other chemical parameters can be used to diagnoses muscle–eye–brian disease. The usage of biopsy includes:
Histological analysis
Direct examination of muscular tissues can proof muscle dystrophy, which can support the potential diagnosis of MEB. Patients are found to have myofibrils round in shape and in markedly various diameters, later nuclei, regenerating fibers and angulated fibers as a result from atrophy.[17][23]
Enzymatic assay
The mutation of MEB involves the malfunction of O-mannosyl ß-1,2-N-acetylgucosaminyltransfersase 1. By measuring the enzymatic activity of this protein, the presence of MEB can be affirmed.[17] Fibroblast and lymphoblasts are chosen to be the assayed participants.[24]
Immunochemistry
Western-blot (immunoblot) can be used to detect the O-mannosyl ß-1,2-N-acetylgucosaminyltransfersase 1 for diagnosis.[23][25] It helps to evaluate the glycosylation state of the protein which it supposed to do.
Difficulties
As a subtype of muscular dystrophy-dystroglycanopathy, MEB is often confused with other sub-type including Walker–Warburg syndrome and Fukuyama congenital muscular dystrophy.[17][26] All these 3 diseases share similar clinical presentation and are classified as the Type A(severe).[26] Overlapped findings contain clinical presentation and immunochemistry result.[27] The diagnosis has to make distinction among them. The decisive evidences for MEB are:
| MEB | WWS | FCMD | |
| Epidemiology[17] | Finland | Worldwide | Japan |
| Characteristic result of physical examination | |||
| Ocular abnormalities[28] | Progressive myopia with retinal degeneration | Severe malformations | Simple myopia and cataract without structural change |
| Common contractures location [17] | Elbow and knee | Elbow | Elbow, knee, ankle and hip |
| Characteristic result of genetic test | |||
| Major defected gene contributor [17] | POMGNT1 | POMT1, POMT2 | FKTN |
| Minor defected gene contributor[29][30][31] | FKRP, FKTN | FKRP, FCMD, LARGE, ISPD | -- |
| Characteristic result of MRI | |||
| Cerebral cortex[32] | Diffused dysplasia | Diffused cobblestone lissencephaly | Frontal polymicrogyria |
| White matter [32] | Uneven T1 and T2 prolongation | No myelin in cerebrum or cerebellum | Delayed cerebral myelination |
| Others | |||
| Serum creatine kinase level[8][33][34] | Normal to elevated | Elevated to high level | Elevated |
MEB: muscle–eye–brain disease
WWS: Walker–Warburg syndrome
FCMD: Fukuyama congenital muscular dystrophy
Management
There is no current curative treatment for any form of muscle dystrophies and only symptomatic care is available for the patients.[17]
The corresponding supportive care to symptoms is:[33][34]
| Symptom | Corresponding support treatment |
| Contractures | Physiological therapy and tendon-release surgery if needed |
| Scoliosis | Bracing, splinting and corrective surgery such as spinal fixation |
| Muscle weakness | Leg braces, wheelchair, occupation therapy |
| Respiratory problem | Regular monitoring by spirometry, noninvasive nighttime ventilation, tracheostomy, assisted coughing techniques |
| Learning disabilities | Specialized education programs |
| Seizures | Anti-seizures medication[35] |
| Vision problems | Wear special eyeglasses [7][35] |
| Nutritional care | Nasogastric tubes for short-term usage, gastrostomy for chronic need |
Prognosis
MEB is a milder type of congenital muscle dystrophies, with a survival up to more than 70 years being possible.[34] The severity of MEB determines its prognosis. Though the prognosis is associated with the progression of symptoms, supportive care enhances the quality of life and also the life expectancy.[29]
Epidemiology
The majority of MEB occurrence is reported in the Finnish population. It is estimated to affect 1 in 50,000 newborns in Finland.[15] MEB is also identified outside of Finland, there were cases of suspected MEB in Japan and Korea.[10] However, worldwide distribution is unclear.[10] According to the European Union, the estimated prevalence of MEB in Europe is 0.12 per 100000.[41]
| Carrier rates [15] | ||
| Ethnicity | Detection Rate | Carrier Frequency |
| Finnish | >99% | 1 in 50 |
| General Population | 88% | 1 in 500 |
The frequency of being a carrier of MEB is 1 in 50 in Finland, carriers for MEB commonly shows no signs or symptoms. However, their offsprings will have a higher chance to be affected by MEB.[15]
History
MEB was first discovered in Finland. In 1978, a patient from Finland showed symptoms including congenital muscular weakness, severe myopia, glaucoma, optical malformation, intellectual disability, retinal hypoplasia, etc.[36] In 1980, 14 more people with similar symptoms were identified in Finland.[37] Similar cases were also published in 1989.[38] The disease was found in Dutch in 1992, 6 people were affected coming from 4 families.[39] After that, more cases were reported outside the Finnish population.
MEB is phenotypically similar to the Walker–Warburg syndrome (WWS), both disorders are congenital muscular dystrophy. In 1990, Santavuori argued to distinct MEB from WWS, since MEB is specifically involving muscle weakness and there is a relatively long survival for MEB patients.[40] In the same year, Dobyns further examined the relation of WWS and MEB.[41]
In 2001, the cause of MEB was first demonstrated as the mutations in the POMGNT1 gene, causing loss of its function.[42]
References
- "Muscle eye brain disease | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 27 October 2019.
- "Muscle eye brain disease | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2019-03-27.
- Fahnehjelm, Kristina Teär; Ygge, Jan; Engman, Mona-Lisa; Mosskin, Mikael; Santavuori, Pirkko; Malm, Gunilla (2001). "A child with Muscle-Eye-Brain disease". Acta Ophthalmologica Scandinavica. 79 (1): 72–75. doi:10.1034/j.1600-0420.2001.079001072.x. ISSN 1600-0420. PMID 11167293.
- Haltia, M.; Leivo, I.; Somer, H.; Pihko, H.; Paetau, A.; Kivelä, T.; Tarkkanen, A.; Tomé, F.; Engvall, E. (1997). "Muscle—eye—brain disease: A neuropathological study". Annals of Neurology. 41 (2): 173–180. doi:10.1002/ana.410410208. ISSN 1531-8249. PMID 9029066. S2CID 32029963.
- Shenoy, Anant M; Markowitz, Jennifer A; Bonnemann, Carsten G; Krishnamoorthy, Kalpathy; Bossler, Aaron D; Tseng, Brian S (March 2010). "Muscle–Eye–Brain Disease". Journal of Clinical Neuromuscular Disease. 11 (3): 124–126. doi:10.1097/CND.0b013e3181c5054d. ISSN 1522-0443. PMC 2925645. PMID 20215985.
- Diesen, C (2004-10-01). "POMGnT1 mutation and phenotypic spectrum in muscle–eye–brain disease". Journal of Medical Genetics. 41 (10): e115. doi:10.1136/jmg.2004.020701. ISSN 1468-6244. PMC 1735594. PMID 15466003.
- Pihko, Helena; Lappi, Marjatta; Raitta, Christina; Sainio, Kimmo; Valanne, Leena; Somer, Hannu; Santavuori, Pirkko (January 1995). "Ocular findings in muscle–eye–brain (MEB) disease: a follow-up study". Brain and Development. 17 (1): 57–61. doi:10.1016/0387-7604(94)00101-3. ISSN 0387-7604. PMID 7762765. S2CID 33926340.
- Falsaperla, Raffaele; Praticò, Andrea D.; Ruggieri, Martino; Parano, Enrico; Rizzo, Renata; Corsello, Giovanni; Vitaliti, Giovanna; Pavone, Piero (2016-08-31). "Congenital muscular dystrophy: from muscle to brain". Italian Journal of Pediatrics. 42 (1): 78. doi:10.1186/s13052-016-0289-9. ISSN 1824-7288. PMC 5006267. PMID 27576556.
- "O52 Visual evoked potentials and electroretinography in brain-dead patients". Neurophysiologie Clinique/Clinical Neurophysiology. 20: 18s. August 1990. doi:10.1016/s0987-7053(05)80490-3. ISSN 0987-7053. S2CID 53160311.
- Taniguchi, K. (2003-03-01). "Worldwide distribution and broader clinical spectrum of muscle–eye–brain disease". Human Molecular Genetics. 12 (5): 527–534. doi:10.1093/hmg/ddg043. ISSN 1460-2083. PMID 12588800.
- Shenoy, Anant M; Markowitz, Jennifer A; Bonnemann, Carsten G; Krishnamoorthy, Kalpathy; Bossler, Aaron D; Tseng, Brian S (March 2010). "Muscle–Eye–Brain Disease". Journal of Clinical Neuromuscular Disease. 11 (3): 124–126. doi:10.1097/CND.0b013e3181c5054d. ISSN 1522-0443. PMC 2925645. PMID 20215985.
- Raducu, Madalina; Cotarelo, Rocío P.; Simón, Rogelio; Camacho, Ana; Rubio-Fernández, Marcos; Hernández-Laín, Aurelio; Cruces, Jesús (February 2014). "Clinical Features and Molecular Characterization of a Patient with Muscle–Eye–Brain Disease: A Novel Mutation in the POMGNT1 Gene". Journal of Child Neurology. 29 (2): 289–294. doi:10.1177/0883073813509119. hdl:10261/124385. ISSN 0883-0738. PMID 24282183. S2CID 9209393.
- Santavuori, P.; Leisti, J.; Kruus, S. (1977). "Muscle, Eye and Brain Disease: A New Syndrome". Neuropediatrics. 8 (S 01): 553. doi:10.1055/s-0028-1091594. ISSN 0174-304X.
- Reference, Genetics Home. "POMGNT1 gene". Genetics Home Reference. Retrieved 2019-03-27.
- "Muscle-eye brain disease - Gene : POMGNT1". GeneAware. Retrieved April 30, 2020.
{{cite web}}: CS1 maint: url-status (link) - Strahl, Sabine; Lommel, Mark (2009-08-01). "Protein O-mannosylation: Conserved from bacteria to humans". Glycobiology. 19 (8): 816–828. doi:10.1093/glycob/cwp066. ISSN 0959-6658. PMID 19429925.
- S, Gosal Gurinder; H, Shah Hitesh (March 2011). "Muscle–Eye–Brain Disease; a Rare Form of Syndromic Congenital Muscular Dystrophy". Malaysian Orthopaedic Journal. 5 (1): 67–70. doi:10.5704/moj.1103.016. ISSN 1985-2533.
- A, Diesen, C Saarinen, A Pihko, H Rosenlew, C Cormand, B Dobyns, W Dieguez, J Valanne, L Joensuu, T Lehesjoki (October 2004). "POMGnT1 mutation and phenotypic spectrum in muscle–eye–brain disease". Journal of Medical Genetics. BMJ Group. 41 (10): e115. doi:10.1136/jmg.2004.020701. OCLC 679802406. PMC 1735594. PMID 15466003.
- Saredi, S.; Ardissone, A.; Ruggieri, A.; Mottarelli, E.; Farina, L.; Rinaldi, R.; Silvestri, E.; Gandioli, C.; D'Arrigo, S. (July 2012). "Novel POMGNT1 point mutations and intragenic rearrangements associated with muscle–eye–brain disease". Journal of the Neurological Sciences. 318 (1–2): 45–50. doi:10.1016/j.jns.2012.04.008. ISSN 0022-510X. PMC 3405532. PMID 22554691.
- Balci, Burcu; Morris-Rosendahl, Deborah J.; Çelebi, Asli; Talim, Beril; Topalo??lu, Haluk; Dinçer, Pervin (2006). "Prenatal diagnosis of muscle–eye–brain disease". Prenatal Diagnosis. 27 (1): 51–54. doi:10.1002/pd.1622. ISSN 0197-3851. PMID 17154333. S2CID 44662331.
- Valanne, L.; Pihko, H.; Katevuo, K.; Karttunen, P.; Somer, H.; Santavuori, P. (August 1994). "MRI of the brain in muscle–eye–brain (MEB) disease". Neuroradiology. 36 (6): 473–476. doi:10.1007/BF00593687. ISSN 0028-3940. PMID 7991095. S2CID 19829870.
- Millichap, J. J.; Nguyen, T.; Ryan, M. E. (2010-05-31). "Teaching NeuroImages: Prenatal MRI of muscle–eye–brain disease". Neurology. 74 (22): e101. doi:10.1212/wnl.0b013e3181e0f84b. ISSN 0028-3878. PMID 20513809.
- Raducu, Madalina; Cotarelo, Rocío P.; Simón, Rogelio; Camacho, Ana; Rubio-Fernández, Marcos; Hernández-Laín, Aurelio; Cruces, Jesús (2013-11-25). "Clinical Features and Molecular Characterization of a Patient with Muscle–Eye–Brain Disease" (PDF). Journal of Child Neurology. 29 (2): 289–294. doi:10.1177/0883073813509119. hdl:10261/124385. ISSN 0883-0738. PMID 24282183. S2CID 9209393.
- Vajsar, Jiri; Zhang, Wenli; Dobyns, William B.; Biggar, Doug; Holden, Kenton R.; Hawkins, Cynthia; Ray, Peter; Olney, Ann H.; Burson, Catherine M. (February 2006). "Carriers and patients with muscle–eye–brain disease can be rapidly diagnosed by enzymatic analysis of fibroblasts and lymphoblasts". Neuromuscular Disorders. 16 (2): 132–136. doi:10.1016/j.nmd.2005.11.012. ISSN 0960-8966. PMID 16427280. S2CID 21928381.
- Geis, Tobias; Marquard, Klaus; Rödl, Tanja; Reihle, Christof; Schirmer, Sophie; von Kalle, Thekla; Bornemann, Antje; Hehr, Ute; Blankenburg, Markus (2013-09-20). "Homozygous dystroglycan mutation associated with a novel muscle–eye–brain disease-like phenotype with multicystic leucodystrophy". Neurogenetics. 14 (3–4): 205–213. doi:10.1007/s10048-013-0374-9. ISSN 1364-6745. PMID 24052401. S2CID 15027740.
- Amberger, Joanna; Bocchini, Carol; Hamosh, Ada (2011-04-05). "A new face and new challenges for Online Mendelian Inheritance in Man (OMIM®)". Human Mutation. 32 (5): 564–567. doi:10.1002/humu.21466. ISSN 1059-7794. PMID 21472891.
- Bertini, Enrico; D'Amico, Adele; Gualandi, Francesca; Petrini, Stefania (December 2011). "Congenital Muscular Dystrophies: A Brief Review". Seminars in Pediatric Neurology. 18 (4): 277–288. doi:10.1016/j.spen.2011.10.010. PMC 3332154. PMID 22172424.
- Cormand, Bru; Avela, Kristiina; Pihko, Helena; Santavuori, Pirkko; Talim, Beril; Topaloglu, Haluk; de la Chapelle, Albert; Lehesjoki, Anna-Elina (January 1999). "Assignment of the Muscle–Eye–Brain Disease Gene to 1p32-p34 by Linkage Analysis and Homozygosity Mapping". The American Journal of Human Genetics. 64 (1): 126–135. doi:10.1086/302206. ISSN 0002-9297. PMC 1377710. PMID 9915951.
- Wang, Ching H.; Bonnemann, Carsten G.; Rutkowski, Anne; Sejersen, Thomas; Bellini, Jonathan; Battista, Vanessa; Florence, Julaine M.; Schara, Ulrike; Schuler, Pamela M. (December 2010). "Consensus Statement on Standard of Care for Congenital Muscular Dystrophies". Journal of Child Neurology. 25 (12): 1559–1581. doi:10.1177/0883073810381924. ISSN 0883-0738. PMC 5207780. PMID 21078917.
- Kang, P. B.; Morrison, L.; Iannaccone, S. T.; Graham, R. J.; Bonnemann, C. G.; Rutkowski, A.; Hornyak, J.; Wang, C. H.; North, K. (2015-03-31). "Evidence-based guideline summary: Evaluation, diagnosis, and management of congenital muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine". Neurology. 84 (13): 1369–1378. doi:10.1212/WNL.0000000000001416. ISSN 0028-3878. PMC 4388744. PMID 25825463.
- Sparks, Susan E.; Quijano-Roy, Susana; Harper, Amy; Rutkowski, Anne; Gordon, Erynn; Hoffman, Eric P.; Pegoraro, Elena (1993). "Congenital Muscular Dystrophy Overview – ARCHIVED CHAPTER, FOR HISTORICAL REFERENCE ONLY". In Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.). Congenital Muscular Dystrophy Overview. GeneReviews®. University of Washington, Seattle. PMID 20301468. Retrieved 2019-03-27.
- Barkovich, A. J. (September 1998). "Neuroimaging manifestations and classification of congenital muscular dystrophies". AJNR. American Journal of Neuroradiology. 19 (8): 1389–1396. ISSN 0195-6108. PMC 8338698. PMID 9763366.
- Kirschner, Janbernd (2013), "Congenital muscular dystrophies", Pediatric Neurology Part III, Handbook of Clinical Neurology, vol. 113, Elsevier, pp. 1377–1385, doi:10.1016/b978-0-444-59565-2.00008-3, ISBN 9780444595652, PMID 23622361, retrieved 2019-03-27
- Horrocks, I.; Muntoni, F.; Longman, C.; Joseph, S. (2014-10-01). "G.P.315: Cases of normal to mildly elevated creatine kinase in muscle–eye–brain disease patients and delay in diagnosis". Neuromuscular Disorders. 24 (9): 916. doi:10.1016/j.nmd.2014.06.405. ISSN 0960-8966. S2CID 53300651.
- Myriad Women's Health https://myriadwomenshealth.com/2014/05/hello-world/. Retrieved 2019-03-27.
{{cite web}}: Missing or empty|title=(help) - RAITTA, CHRISTINA; LAMMINEN, MAIJA; SANTAVUORI, PIRKKO; LEISTI, JAAKKO (2009-05-27). "Ophthalmological Findings in a New Syndrome with Muscle, Eye and Brain Involvement". Acta Ophthalmologica. 56 (3): 465–472. doi:10.1111/j.1755-3768.1978.tb05700.x. ISSN 1755-375X. PMID 581135. S2CID 28078013.
- W., Eriksson, Aldur (1980). Population structure and genetic disorders : Mariehamm, Åland Islands, Finland, August 1978. Academic Pr. ISBN 0122414500. OCLC 313921203.
- Santavuori, P.; Somer, H.; Sainio, K.; Rapola, J.; Kruus, S.; Nikitin, T.; Ketonen, L.; Leisti, J. (1989). "Muscle–eye–brain disease (MEB)". Brain & Development. 11 (3): 147–153. doi:10.1016/S0387-7604(89)80088-9. ISSN 0387-7604. PMID 2751061. S2CID 4702708.
- Leyten, Q. H.; Gabreëls, F. J.; Renier, W. O.; Renkawek, K.; ter Laak, H. J.; Mullaart, R. A. (December 1992). "Congenital muscular dystrophy with eye and brain malformations in six Dutch patients". Neuropediatrics. 23 (6): 316–320. doi:10.1055/s-2008-1071365. ISSN 0174-304X. PMID 1491751.
- Santavuori, P.; Pihko, H.; Sainio, K.; Lappi, M.; Somer, H.; Haltia, M.; Raitta, C.; Ketonen, L.; Leisti, J. (July 1990). "Muscle–eye–brain disease and Walker–Warburg syndrome". American Journal of Medical Genetics. 36 (3): 371–374. doi:10.1002/ajmg.1320360334. ISSN 0148-7299. PMID 2363444.
- Dobyns, William B.; Pagon, Roberta A.; Curry, Cynthia J. R.; Greenberg, Frank (July 1990). "Response to Santavuori et al. regarding Walker–Warburg syndrome and muscle–eye–brain disease". American Journal of Medical Genetics. 36 (3): 373–374. doi:10.1002/ajmg.1320360335. ISSN 0148-7299.
- Yoshida, A.; Kobayashi, K.; Manya, H.; Taniguchi, K.; Kano, H.; Mizuno, M.; Inazu, T.; Mitsuhashi, H.; Takahashi, S. (November 2001). "Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1". Developmental Cell. 1 (5): 717–724. doi:10.1016/S1534-5807(01)00070-3. ISSN 1534-5807. PMID 11709191.