Holoprosencephaly
Holoprosencephaly (HPE) is a cephalic disorder in which the prosencephalon (the forebrain of the embryo) fails to develop into two hemispheres, typically occurring between the 18th and 28th day of gestation.[1] Normally, the forebrain is formed and the face begins to develop in the fifth and sixth weeks of human pregnancy. The condition also occurs in other species.
| Holoprosencephaly | |
|---|---|
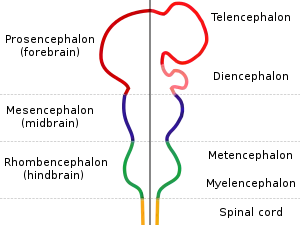 | |
| Diagram depicting the main subdivisions of the embryonic vertebrate brain. | |
| Specialty | Medical genetics |
Holoprosencephaly is estimated to occur in approximately 1 in every 250 conceptions[1] and most cases are not compatible with life and result in fetal death in utero due to deformities to the skull and brain.[2] However, holoprosencephaly is still estimated to occur in approximately 1 in every 8,000 live births.[3]
When the embryo's forebrain does not divide to form bilateral cerebral hemispheres (the left and right halves of the brain), it causes defects in the development of the face and in brain structure and function.
The severity of holoprosencephaly is highly variable. In less severe cases, babies are born with normal or near-normal brain development and facial deformities that may affect the eyes, nose, and upper lip.[4]
Signs and symptoms
Symptoms of holoprosencephaly range from mild (no facial/organ defects, anosmia, or only a single central incisor) to moderate to severe (cyclopia). The symptoms are dependent upon the classification type.[3]
There are four classifications of holoprosencephaly as well as a mild "microform".
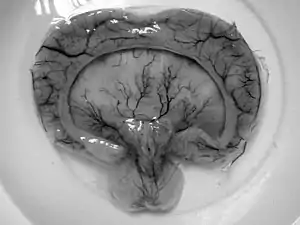
- Alobar
- Most severe form includes formation of synophthalmia (a single central eye), proboscis, and severe impairment.[3]
- Semilobar
- Can present with severely decreased distance between eyes, a flat nasal bridge, eye defects, cleft lip and palate, and severe impairment.[3]
- Lobar
- Can present with decreased distance between eyes, a flat nasal bridge, closely spaced nostrils, mental and locomotion delays may be present.[3]
- Syntelencephaly or middle interhemispheric variant of holoprosencephaly (MIHV)
- Mild phenotypic presentation which can present with flat nasal bridge, metopic prominence, shallow philtrum, and possible mental and locomotion delays.[5]
- "Microform"
- Mild phenotypic presentation with reduced distance between eyes, sharp nasal bridge, single maxillary central incisor.[3]
Diagnosis
Holoprosencephaly is typically diagnosed during fetal development when there are abnormalities found on fetal brain imaging, however it can also be diagnosed after birth. The Protocol for diagnosis includes neuroimaging (Ultrasound or fetal MRI prior to birth or Ultrasound, MRI or CT post birth), syndrome evaluation, cytogenetics, molecular testing, and genetic counseling.[3]
There are four classifications of holoprosencephaly as well as a “microform".[3] These classifications can be distinguished by their anatomical differences.[1]
- Alobar holoprosencephaly
- Small single forebrain ventricle
- No interhemispheric division
- Absence of olfactory bulbs and tracts
- Absence of corpus callosum
- Non separation of deep gray nuclei[1]
- Semilobar holoprosencephaly
- Rudimentary cerebral lobes
- Incomplete interhemispheric division
- Absence or hypoplasia of olfactory bulbs and tracts
- Absence of corpus callosum
- Varying non separation of deep gray nuclei[1]
- Lobar holoprosencephaly
- Fully-developed cerebral lobes
- Distinct interhemispheric division
- Midline continuous frontal neocortex
- Absent, hypoplasic or normal corpus callosum
- Separation of deep gray nuclei[1]
- Syntelencephaly, or middle interhemispheric variant of holoprosencephaly (MIHV)
- Failure of separation of the posterior frontal and parietal lobes
- Callosal genu and splenium normally formed
- Absence of corpus callosum
- Hypothalamus and lentiform nuclei normally separated
- Heterotopic gray matter[1]
- Microform
- Subtle defects of corpus callosum
- Subtle midline brain defects[3]
Causes
In Holoprosencephaly, the neural tube fails to segment, resulting in incomplete separation of the prosencephalon at the fifth week of gestation.[6]
The exact cause(s) of HPE are yet to be determined. Mutations in the gene encoding the SHH protein, which is involved in the development of the central nervous system (CNS), can cause holoprosencephaly.[7][8][9] In other cases, it often seems that there is no specific cause at all.[10]
Genetics
Armand Marie Leroi describes the cause of cyclopia as a genetic malfunctioning during the process by which the embryonic brain is divided into two.[11] Only later does the visual cortex take recognizable form, and at this point an individual with a single forebrain region will be likely to have a single, possibly rather large, eye (at such a time, individuals with separate cerebral hemispheres would form two eyes).
Increases in expression of such genes as Pax-2, as well as inhibition of Pax-6, from the notochord have been implicated in normal differentiation of cephalic midline structures. Inappropriate expression of any of these genes may result in mild to severe forms of holoprosencephaly. Other candidate genes have been located, including the SHH (holoprosencephaly type 3 a.k.a. HPE3), TGIF, ZIC2, SIX3[12] and BOC genes.[13]
Although many children with holoprosencephaly have normal chromosomes, specific chromosomal abnormalities have been identified in some patients (trisomy of chromosome 13, also known as Patau syndrome). There is evidence that in some families, HPE is inherited (autosomal dominant as well as autosomal or X-linked recessive inheritance).[14][15][16] Features consistent with familial transmission of the disease (e.g., a single central maxillary incisor) should be carefully assessed in parents and family members.[17]
Non-genetic factors
Numerous possible risk factors have been identified, including gestational diabetes, transplacental infections (the "TORCH complex"), first trimester bleeding, and a history of miscarriage.[10][18] As well, the disorder is found twice as often in female babies.[18] However, there appears to be no correlation between HPE and maternal age.[18]
There is evidence of a correlation between HPE and the use of various drugs classified as being potentially unsafe for pregnant and lactating mothers. These include insulin, birth control pills, aspirin, lithium, thorazine, retinoic acid, and anticonvulsants.[18] There is also a correlation between alcohol consumption and HPE, along with nicotine, the toxins in cigarettes and toxins in cigarette smoke when used during pregnancy.[18]
Prognosis
HPE is not a condition in which the brain deteriorates over time. Although serious seizure disorders, autonomic dysfunction, complicated endocrine disorders and other life-threatening conditions may sometimes be associated with HPE, the mere presence of HPE does not mean that these serious problems will occur or develop over time without any previous indication or warning. These abnormalities are usually recognized shortly after birth or early in life and only occur if areas of the brain controlling those functions are fused, malformed or absent.[4]
Prognosis is dependent upon the degree of fusion and malformation of the brain, as well as other health complications that may be present.
The more severe forms of encephalopathy are usually fatal. This disorder consists of a spectrum of defects, malformations and associated abnormalities. Disability is based upon the degree in which the brain is affected. Moderate to severe defects may cause intellectual disability, spastic quadriparesis, athetoid movements, endocrine disorders, epilepsy and other serious conditions; mild brain defects may only cause learning or behavior problems with few motor impairments.
Seizures may develop over time with the highest risk before 2 years of age and the onset of puberty. Most are managed with one medication or a combination of medications. Typically, seizures that are difficult to control appear soon after birth, requiring more aggressive medication combinations/doses.
Most children with HPE are at risk of having elevated blood sodium levels during moderate-severe illnesses, that alter fluid intake/output, even if they have no previous diagnosis of diabetes insipidus or hypernatremia.
References
- Dubourg, Christèle; Bendavid, Claude; Pasquier, Laurent; Henry, Catherine; Odent, Sylvie; David, Véronique (2007-02-02). "Holoprosencephaly". Orphanet Journal of Rare Diseases. 2 (1): 8. doi:10.1186/1750-1172-2-8. ISSN 1750-1172. PMC 1802747. PMID 17274816.
- "Holoprosencephaly Information Page". National Institute of Neurological Disorders and Stroke. National Institutes of Health, U.S. Department of Health & Human Services.
- Raam, Manu S; Solomon, Benjamin D; Muenke, Maximilian (June 2011). "Holoprosencephaly: A Guide to Diagnosis and Clinical Management". Indian Pediatrics. 48 (6): 457–466. doi:10.1007/s13312-011-0078-x. ISSN 0019-6061. PMC 4131946. PMID 21743112.
- Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V (February 2007). "Holoprosencephaly". Orphanet Journal of Rare Diseases. 2 (1): 8. doi:10.1186/1750-1172-2-8. PMC 1802747. PMID 17274816.
- Rajalakshmi, P. Prathiba; Gadodia, Ankur; Priyatharshini, P. (2015). "Middle interhemispheric variant of holoprosencephaly: A rare midline malformation". Journal of Pediatric Neurosciences. 10 (3): 244–246. doi:10.4103/1817-1745.165678. ISSN 1817-1745. PMC 4611894. PMID 26557166.
- Winter, Thomas C.; Kennedy, Anne M.; Woodward, Paula J. (2015-01-01). "Holoprosencephaly: A Survey of the Entity, with Embryology and Fetal Imaging". RadioGraphics. 35 (1): 275–290. doi:10.1148/rg.351140040. ISSN 0271-5333. PMID 25590404.
- Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA (October 1996). "Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function". Nature. 383 (6599): 407–413. Bibcode:1996Natur.383..407C. doi:10.1038/383407a0. PMID 8837770. S2CID 4339131.
- Muenke M, Beachy PA (June 2000). "Genetics of ventral forebrain development and holoprosencephaly". Current Opinion in Genetics & Development. 10 (3): 262–269. doi:10.1016/s0959-437x(00)00084-8. PMID 10826992.
- Rash BG, Grove EA (October 2007). "Patterning the dorsal telencephalon: a role for sonic hedgehog?". The Journal of Neuroscience. 27 (43): 11595–11603. doi:10.1523/jneurosci.3204-07.2007. PMC 6673221. PMID 17959802.
- The Carter Centers for Brain Research in Holoprosencephaly and Related Malformations. "About Holoprosencephaly". Archived from the original on 2009-05-14.
- Leroi AM (2003). Mutants : on the form, varieties and errors of the human body. London: HarperCollins. ISBN 978-0-00-653164-7.
- "The Carter Center for Research in holoprosencephaly". Archived from the original on 2008-11-21.
- Hong M, Srivastava K, Kim S, Allen BL, Leahy DJ, Hu P, et al. (November 2017). "BOC is a modifier gene in holoprosencephaly". Human Mutation. 38 (11): 1464–1470. doi:10.1002/humu.23286. PMC 5673120. PMID 28677295.
- Singh S, Tokhunts R, Baubet V, Goetz JA, Huang ZJ, Schilling NS, et al. (February 2009). "Sonic hedgehog mutations identified in holoprosencephaly patients can act in a dominant negative manner". Human Genetics. 125 (1): 95–103. doi:10.1007/s00439-008-0599-0. PMC 2692056. PMID 19057928.
- Tekendo-Ngongang C, Muenke M, Kruszka P (1993). "Holoprosencephaly Overview". In Adam MP, Ardinger HH, Pagon RA, Wallace SE (eds.). GeneReviews®. Seattle (WA): University of Washington, Seattle. PMID 20301702. Retrieved 2020-09-01.
- Nanni L, Ming JE, Bocian M, Steinhaus K, Bianchi DW, Die-Smulders C, et al. (December 1999). "The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly". Human Molecular Genetics. 8 (13): 2479–2488. doi:10.1093/hmg/8.13.2479. PMID 10556296.
- Nanni L, Ming JE, Du Y, Hall RK, Aldred M, Bankier A, Muenke M (July 2001). "SHH mutation is associated with solitary median maxillary central incisor: a study of 13 patients and review of the literature". American Journal of Medical Genetics. 102 (1): 1–10. doi:10.1002/1096-8628(20010722)102:1<1::aid-ajmg1336>3.0.co;2-u. PMID 11471164.
- Croen LA, Shaw GM, Lammer EJ (February 2000). "Risk factors for cytogenetically normal holoprosencephaly in California: a population-based case-control study". American Journal of Medical Genetics. 90 (4): 320–325. doi:10.1002/(SICI)1096-8628(20000214)90:4<320::AID-AJMG11>3.0.CO;2-8. PMID 10710231.