Saethre–Chotzen syndrome
Saethre–Chotzen syndrome (SCS), also known as acrocephalosyndactyly type III, is a rare congenital disorder associated with craniosynostosis (premature closure of one or more of the sutures between the bones of the skull). This affects the shape of the head and face, resulting in a cone-shaped head and an asymmetrical face. Individuals with SCS also have droopy eyelids (ptosis), widely spaced eyes (hypertelorism), and minor abnormalities of the hands and feet (syndactyly).[2] Individuals with more severe cases of SCS may have mild to moderate intellectual or learning disabilities. Depending on the level of severity, some individuals with SCS may require some form of medical or surgical intervention.[3] Most individuals with SCS live fairly normal lives, regardless of whether medical treatment is needed or not.[2]
| Saethre–Chotzen syndrome | |
|---|---|
| Other names |
|
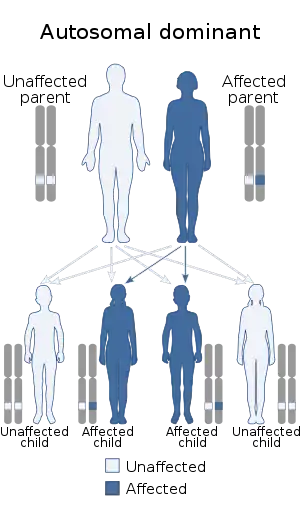 | |
| This condition is inherited in an autosomal dominant manner | |
| Specialty | Rheumatology |
Signs and symptoms
SCS presents in a variable fashion. The majority of individuals with SCS are moderately affected, with uneven facial features and a relatively flat face due to underdeveloped eye sockets, cheekbones, and lower jaw. In addition to the physical abnormalities, people with SCS also experience growth delays, which results in a relatively short stature. Although, most individuals with SCS are of normal intelligence, some individuals may have mild to moderate mental delays. More severe cases of SCS, with more serious facial deformities, occurs when multiple cranial sutures close prematurely.[2]
Cranial defects

- Flat, asymmetric head and face[3]
- Head is typically cone-shaped (acrocephaly) or flat (brachycephaly) but can also be long and narrow (dolichocephaly)[4]
- Head is short from front to back[5]
- Lopsided face[4]
- Low-set hairline causing forehead to appear tall and wide[5]
Defects of the hands and feet
- Webbing (syndactyly) between the second and third finger and between the second and third toes[2]
- Short fingers and toes (brachydactyly)[4]
- Broad thumb and/or a broad hallux (big toe) with a valgus deformity (outward angulation of the distal segment of a bone/joint)[6]
- Hands have a single palmer flexion crease[3]
Ocular defects

- Unevenly positioned eyes that may be crossed (strabismus) or wide-set (hypertelorism)[5]
- Vision problems due to abnormal facial anatomy, which causes mechanical disturbances of the extraocular muscles, resulting in strabismus (crossed eyes)[3]
- Tear duct stenosis (narrowing of the tear duct)[3]
- Drooping eyelids (ptosis)[3]
- Downward slanting palpebral fissures (separation between upper and lower eyelids)[3]
- Nearsightedness (myopia)[4]
- Epicanthal folds (skin folds of the upper eyelid covering the inner corner of the eye)[6]
- Blepharophimosis (bilateral ptosis with reduced size of eyelid)[6]
- Optic atrophy[6]
- Refractory errors[6]
Ear, nose, and mouth defects
- Small, low-set ears that may be rotated somewhat backwards and has a prominent (bulging) pinna[4]
- Beaked nose (slightly bent downward at tip) that is slightly off center and contains a deviated septum[2]
- Malocclusion associated with dental abnormalities including enamel hypoplasia (thin enamel due to incomplete formation), hyperdontia (extra teeth), and peg teeth (small, abnormally shaped teeth)[6]
- Cleft palate with high arch[6]
Less common defects
- Short stature[7]
- Vertebral fusions[7]
- Congenital heart problems[6]
- Speech problems[8]
- Anal atresia (malformed rectum)[3]
- Undescended testes (cryptorchidism)[4]
- Renal (kidney) abnormalities[4]
- Personality disorders[6]
Causes
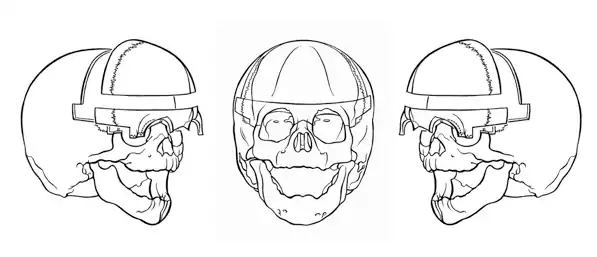
Craniosynostosis

The cranium consists of three main sections including the base of the cranium (occipital bone), the face (frontal bone), and the top (parietal bones) and sides (temporal bone) of the head. Most of the bones of the cranium are permanently set into place prior to birth. However, the temporal and parietal bones are separated by sutures, which remain open, allowing the head to slightly change in shape during childbirth. The cranial sutures eventually close within the first couple of years following birth, after the brain has finished growing.[2]
In individuals with SCS, the coronal suture separating the frontal bones from the parietal bones, closes prematurely (craniosynostosis), occasionally even before birth. If the coronal suture closes asymmetrically or unilaterally, then the face and forehead will form unevenly, from side-to-side. People with SCS have pointy, tower-like heads because their brain is growing faster than their skull, resulting in increased intracranial pressure (ICP) and causing the top of the head and/or forehead to bulge out to allow for brain growth. The face appears uneven, particularly in the areas of the eyes and cheeks, and the forehead appears wide and tall.[2]
Because of the abnormal forehead, there is less space for the normal facial features to develop. This results in shallow eye sockets and flat cheekbones. The shallow eye sockets make the eyes more prominent or bulging and cause the eyes to be more separated than normal (hypertelorism). The underdeveloped eye sockets, cheekbones, and lower jaw cause the face to appear flat. Furthermore, the minor downward slant of the eyes along with the drooping eyelids (ptosis) adds to the overall unevenness of the face.[2]
Genetics

SCS is typically inherited as an autosomal dominant trait. However, on occasion, children with a microdeletion of 7p21 (chromosome containing the locus responsible for SCS) develop new abnormalities and typically show significant neurological abnormalities. An increased parental age may play a role in the development of new mutations and abnormalities.[3]
Linkage analysis and chromosomal rearrangement revealed the cause of SCS to be mutations in the TWIST gene (twist transcription factor gene) located on chromosome 7p21. The TWIST gene encodes a basic helix-loop-helix (b-HLH) transcription factor that controls head mesenchyme development as the cranial tube forms. More than 35 varying TWIST mutations involving the b-HLH domain of the protein have been identified in people with SCS. The mutations include missense, nonsense, and frameshift deletion/insertion mutations that either shorten or disrupt the b-HLH domain. Most individuals with SCS have a single large deletion in the region 7p21, which contains the region that codes for the TWIST gene.[6]
In searching for the gene responsible for SCS, scientists at Johns Hopkins Children’s Center began studying the TWIST gene because its effects on mice. The TWIST gene in mice, functions in the development of the muscle and skeleton of the face, head, hands, and feet. Mice that were lacking both copies of the TWIST gene were spontaneously aborted prior to birth, and had serious deformities including abnormal limb and head defects and failure of the neural tube to properly close. However, mice with a single copy of the non-working TWIST gene survived. Further examination revealed that these mice had only minor skull, hand, and foot defects similar to those seen in SCS. The mouse TWIST gene is located on chromosome 12 in mice, which corresponds to the short arm of chromosome 7 in humans. With this information, scientists began to isolate and map the human TWIST gene on the short arm of human chromosome 7. They revealed that the human TWIST gene was in the same region that was absent in people with SCS. While looking for different mutations in the human TWIST gene, five different types of mutations were discovered in individuals with SCS. Since none of these mutations were seen in normal individuals who didn't have SCS, this provided enough evidence to conclude that the TWIST gene was the causative agent of SCS1. Researchers also studied the TWIST gene in Drosophila (fruit fly) in order to determine its function. They discovered that in the presence of two TWIST protein molecules combined together, the TWIST gene functions as a DNA transcription factor, meaning it binds to the DNA double-helix at specific locations in order to control which genes are "turned on" or activated. The majority of the identified mutations in the TWIST gene interfere with how the protein attaches to DNA, preventing the activation of other genes that would normally be turned on during fetal development.[2]
Diagnosis
Prenatal diagnosis
Prenatal diagnosis of Saethre-Chotzen Syndrome in high risk pregnancies is doable, but very uncommon and rarely performed. Furthermore, this is only possible if the mutation causing the disease has already been identified within the family genome. There are a few different techniques in which prenatal testing can be carried out. Prenatal testing is usually performed around 15–18 weeks, using amniocentesis to extract DNA from the fetus's cells. Prenatal testing can also be performed during weeks 10–12 using chorionic villus sampling (CVS) to extract DNA from the fetus.[7] Recently, there has been an increased interest in utilizing ultrasound equipment in order to detect fetal skull abnormalities due to immature fusion of the cranial sutures.[4]
Clinical diagnosis
The overall diagnosis of SCS is primarily based on clinical findings and observations based on dysmorphology examination (assessing structural defects) and radiographic evaluation (X-rays, MRIs, and CT scans).[6]
Molecular/genetic diagnosis
A clinical diagnosis of SCS can be verified by testing the TWIST1 gene (only gene in which mutations are known to cause SCS) for mutations using DNA analysis, such as sequence analysis, deletion/duplication analysis, and cytogenetics/ FISH analysis. Sequence analysis of exon 1 (TWIST1 coding region) provides a good method for detecting the frequency of mutations in the TWIST1 gene. These mutations include nonsense, missense, splice site mutation, and intragenic deletions/insertions. Deletion/duplication analysis identifies mutations in the TWIST1 gene that are not readily detected by sequence analysis. Common methods include PCR, multiplex ligation-dependent probe amplification (MLPA), and chromosomal microarray (CMA). Cytogenetic/FISH analysis attaches fluorescently labels DNA markers to a denatured chromosome and is then examined under fluorescent lighting, which reveals mutations caused by translocations or inversions involving 7p21. Occasionally, individuals with SCS have a chromosome translocation, inversion, or ring chromosome 7 involving 7p21 resulting in atypical findings, such as, increased developmental delay.[7] Individuals with SCS, typically have normal brain functioning and rarely have mental impairments. For this reason, if an individual has both SCS and mental retardation, then they should have their TWIST1 gene screened more carefully because this is not a normal trait of SCS.[2] Cytogenetic testing and direct gene testing can also be used to study gene/chromosome defects. Cytogenetic testing is the study of chromosomes to detect gains or losses of chromosomes or chromosome segments using fluorescent in situ hybridization (FISH) and/or comparative genomic hybridization (CGH). Direct gene testing uses blood, hair, skin, amniotic fluid, or other tissues in order to find genetic disorders. Direct gene testing can determine whether an individual has SCS by testing the individual's blood for mutations in the TWIST1 gene.[7]
Differential diagnosis
Genetic testing allows for a definitive diagnosis because it allows similar conditions to be differentiated from each other based on which gene is mutated.[2] The following table contains conditions similar to SCS:
| Condition | Symptoms | Gene |
|---|---|---|
| SCS | Widely spaced eyes, low hairline, drooping eyes, interdigital webbing, deformed ears, crossed eyes, and downward sloping palpebral fissures | TWIST1 |
| Robinow–Sorauf syndrome | Widely spaced eyes, deviated septum, flat skull posterior, deformed ears, crossed eyes, protruding jaw, and duplication of distal phalanx | TWIST1 |
| Muenke syndrome | Widely spaced eyes, enlarged head, hearing loss, flat cheeks, and low-set ears | FGFR3 |
| Crouzon syndrome | Widely spaced eyes, short-broad head, hearing loss, bulging eyes, beaked nose, low-set ears, strabismus, protruding chin, and short humerus and femur | FGFR2 & FGFR3 |
| Pfeiffer syndrome | Widely spaced eyes, underdeveloped jaw, beaked nose, hearing loss, and bulging eyes | FGFR1 & FGFR2 |
| Apert syndrome | Widely spaced eyes, prominent forehead, flat skull posterior, bulging eyes, low-set ears, flat or concave face, short thumb, and webbed fingers | FGFR2 |
| Isolated unilateral coronal synostosis | Only malformation is the premature fusion of sutures; If left untreated, can lead to facial asymmetry resembling SCS | FGFR (any) |
| Baller–Gerold syndrome (BGS) | Short broad head, bulging eyes, flat forehead, poikiloderma, radial deformity with reduced number of digits, underdeveloped or missing thumb and radius, and growth retardation | RECQL4 |
Treatment
The physical abnormalities resulting from SCS are typically mild and only require a minor surgical procedure or no procedure at all. One of the common symptoms of SCS is the development of short (brachydactyly), webbed fingers and broad toes (syndactyly). These characteristics do not cause any problems to the function of the hands or feet, and thus, no medical procedure is required to fix the abnormalities, unless the patient requests it. Webbing of the fingers may affect the base of the fingers, resulting in delayed hand growth during childhood, but this contributes no functional impairments. Sometimes, individuals with SCS develop broad toes because the bones at the ends of the toes are duplicating themselves. This is especially seen in the big toe, but requires no surgical intervention because it doesn't negatively affect the overall function of the foot. Individuals with these toe abnormalities walk normally and can wear normal footwear.[9]
In more severe cases, frequent surgeries and clinical monitoring are required throughout development. A child born with asymmetrical unilateral coronal synostosis should undergo cranioplasty within its first year of life in order to prevent increased intracranial pressure and to prevent progressive facial asymmetry. Cranioplasty is a surgical procedure to correct prematurely fused cranial bones. The surgery acts to reconstruct and reposition the bones and sutures in order to promote the most normal growth.[6] Cranioplasty is necessary in order to continue to grow and is important for two main reasons. First of all, the skull needs to be able to accommodate the growing brain following childbirth, which it can't because the skull doesn't grow as fast as the brain as long as the sutures remain fused. This results in an increase in pressure surrounding the brain and inhibits the brain from growing, causing the individual to experience significant problems, and if left untreated can eventually lead to death. Secondly, cranioplasty may be required for appearance purposes.[7] This is especially the case in individuals with asymmetrical unilateral coronal synostosis, which requires reconstructive surgery of the face and skull. If cranioplasty is not performed, especially in individuals with unilateral coronal synostosis, then facial asymmetry will get worse and worse over time, which is why cranioplasty should be performed as soon as possible.[9]
Surgery may also be required in individuals with vision problems. Vision problems usually arise due to a lack of space in the eye orbit and skull because of the abnormal bone structure of the face. Decreased space may also lead to abnormal or missing tear ducts and nerve damage. Reconstructive surgery is usually required in order to increase cranial space, correct tear duct stenosis, and/or correct ptosis of the eyelids in order to prevent amblyopia (lazy eye).[2]
Midfacial surgery may also be required during early childhood to correct respiratory problems, dental malocclusion, and swallowing difficulties. A cleft palate is also corrected with surgery, and may involve the use of tympanostomy tubes. If needed, an individual will undergo orthognathic treatment and/or orthodontic treatment after facial development is complete.[2] Since hearing loss is frequently associated with SCS, it is recommended that audiology screening persist throughout childhood.[6]
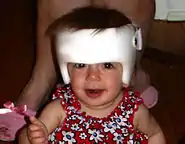
After cranial reconstructive surgery, a child may be required to wear a molding helmet or some other form of head protection until the cranial bones set into place. This typically takes about three months and depends on the child's age and the severity of the condition. Following recovery, individuals with SCS look and act completely normal, so no one would even be able to tell that they have SCS.[10]
Epidemiology
SCS is the most common craniosynostosis syndrome and affects 1 in every 25,000 to 50,000 individuals.[11] It occurs in all racial and ethnic groups, and affects males and females equally.[2] If a parent carries a copy of the SCS gene mutation, then there is a 50% chance their child will also carry a copy of the gene mutation, in which case, the child may or may not show signs of SCS. There is also a 50% chance their child will have two working copies of the gene, and would therefore, not have SCS. If both parents carry a single copy of the SCS gene mutation, then there is a 25% chance their child will have two gene mutation copies (so child would develop severe SCS), a 25% chance their child would have two normal copies of the gene (so would be completely normal), and a 50% chance their child would carry one gene mutation copy and 1 normal copy (so child may or may not display SCS).[2] In rare situations, two normal parents can have a child with SCS due to a de novo mutation. The exact cause of the de novo mutation is unknown, but it doesn't seem to be related to anything that the parents did or didn't do during the pregnancy.[12] SCS due to a de novo mutation is so rare that the proportion of past cases is unknown.[7]
History
In 1931, Haakon Saethre, a Norwegian psychiatrist, described similar characteristics between a mother and her two daughters. They all had long and uneven facial features, low-set hairlines, short fingers, and webbing between the second and third fingers and between the second, third, and fourth toes. A year later in 1932, F. Chotzen, a German psychiatrist, described a father and his two sons as having very similar characteristics as the mother and her daughters, as well as having hearing loss, short stature, and mild mental retardation. Hence, the name Saethre-Chotzen Syndrome was derived from the two scientists, who had separately described the condition without any previous knowledge of the other.[2]
References
- "Children's Health: Saethre Chotzen Syndrome". WebMD. Retrieved November 28, 2012.
- Blanchford, Stacey L (2002). The Gale Encyclopedia of Genetic Disorders. Michigan: Gale Group. pp. 1019–1021. ISBN 9780787656140.
- Allanson, Judith, Cassidy, Suzanne (2010). Management of Genetic Syndromes. New Jersey: John Wiley & Sons, Inc. pp. 230–235. ISBN 9780470191415.
- Wynbrandt, James (2008). Genetic Disorders and Birth Defects. New York: Facts on File, Inc. pp. 340. ISBN 9780816063963.
- "Saethre-Chotzen Syndrome". International Craniofacial Institute. Retrieved Oct 28, 2012.
- Clauser L, Galie M. "Saethre-Chotzen Syndrome" (PDF). Orphanet. Retrieved Oct 28, 2012.
- Gallagher E, Ratisoontorn C, Cunningham M (1993). "Saethre-Chotzen Syndrome". Saethre Chotzen Syndrome. NCBI. PMID 20301368. Retrieved Oct 28, 2012.
- "Saethre-Chotzen Syndrome". Children's Hospital and Medical Center. Retrieved Oct 25, 2012.
- Anderson, Peter. "Headlines Craniofacial Support" (PDF). Archived from the original (PDF) on June 30, 2012. Retrieved November 27, 2012.
- "Surgical Options for Craniosynostosis". Johns Hopkins Medicine. Retrieved November 28, 2012.
- "Saethre-Chotzen Syndrome". Boston Children's Hospital. Retrieved November 28, 2012.
- "Saethre-Chotzen Syndrome". Seattle Children's Hospital. Retrieved November 28, 2012.