Kalkitoxin
Kalkitoxin, a toxin derived from the cyanobacterium Lyngbya majuscula, induces NMDA receptor mediated neuronal necrosis, blocks voltage-dependent sodium channels, and induces cellular hypoxia by inhibiting the electron transport chain (ETC) complex 1.
| Names | |
|---|---|
| Preferred IUPAC name
(2R)-N-{(3S,5S,6R)-7-[(4R)-4-Ethenyl-4,5-dihydro-1,3-thiazol-2-yl]-3,5,6-trimethylheptyl}-N,2-dimethylbutanamide | |
| Identifiers | |
CAS Number |
|
3D model (JSmol) |
|
| ChemSpider | |
PubChem CID |
|
| UNII | |
CompTox Dashboard (EPA) |
|
InChI
| |
SMILES
| |
| Properties | |
Chemical formula |
C21H38N2OS |
| Molar mass | 366.61 g·mol−1 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
Infobox references | |
Natural sources
Kalkitoxin is an ichthyotoxin, derived from the cyanobacterium Lyngbya majuscula [1] which covers sections of the coral reef.[2] It typically forms mini-blooms[2] and produces several metabolites, such as kalkitoxin, curacin-A and antillatoxin.[1] Kalkitoxin has been found and purified near the coasts of Curaçao[1] and Puerto Rico.[3]
Structure and reactivity
Kalkitoxin is a lipopeptide toxin [4] with a molecular weight of 366.604Da.[5] Its chemical formula is C21H38N2OS.[6] The structure contains two double bonds, a 2,4-disubstituted thiazoline ring system, and an additional carbonyl-group.[6] These four groups each provide a degree of unsaturation, which causes kalkitoxin to have four degrees of unsaturation.[6] The structure contains 5 chiral centers, one of which is due to a substituent of the thiazoline ring, and the other four are due to methine groups along the aliphatic carbon chain,[7] which are tertiary carbon atoms bearing three single carbon bonds and one hydrogen. The four methyl groups (each at a methine chiral center), the structure's overall stereochemistry, and the N-methyl group all contribute to the toxicity of kalkitoxin.[7]
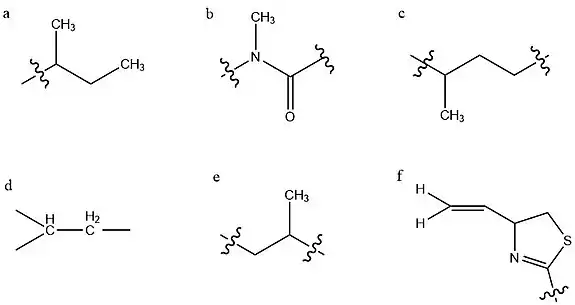
Structure determination
The structure of kalkitoxin was first determined by characterizing six partial structures which were subsequently connected to yield the total structure.[4] This investigation was largely carried out through various NMR experiments. Structure (a) is a sec-butyl group, indicated by characteristic deshielding of its central methine group due to the adjacent carbonyl. Structure (b) contains this carbonyl group, and an adjacent tertiary methylated nitrogen atom, constituting a tertiary amide group. Since this is a tertiary amide, it exists in a cis/trans mixture, which underlies the two conformations of kalkitoxin. Structure (c) is a string of two methylene groups, then a methine group bearing a high-field methyl group. The next two groups identified (d,e) are identical and opposing strings of CH2-CH-CH3, however the left grouping's methylene protons experience greater deshielding, due to their proximity to the adjacent imine. Deshielding is an effect of a nearby electronegative atom withdrawing electron density from a given atom nucleus, eliciting an increased chemical shift as measured by NMR.
The final partial structure consists of a thiazoline ring with a terminal alkene substituent, as determined by electron ionization mass spectrometry (EI-MS) and 13C NMR.[4] The chemical shifts of ring carbons adjacent to the sulfur and nitrogen heteroatoms were compared to 13C NMR data from model compounds. This allowed for the determination of these heteroatoms' locations in the ring, and subsequently the existence of the thiazoline ring itself.[8] With these partial structures established, their connectivity was evaluated via HMBC spectroscopy,[4] a 2D NMR technique which allows for the determination of heteronuclear J-coupling values for nonadjacent carbons and protons. This allows for the spatial relation of specific carbon and hydrogen atoms within a structure to be determined.
Stereochemistry
Kalkitoxin has five chiral centers, one of which is the ring carbon to which the terminal alkene is coordinated, with the remaining four occurring at tertiary carbon atoms along the aliphatic chain originating from the imine nitrogen. The total stereochemistry of natural (+)-kalkitoxin is 3R,7R,8S,10S,2′R.[6] For this determination, 3JCH values by a variation of the HSQMBC pulse technique, a type of HMBC spectroscopy, and 3JHH values by exclusive correlation spectroscopy (E.COSY). These methods use NMR to evaluate the spin-spin coupling constants which directly relate to the dihedral angle of the atoms being analyzed, allowing for the determination of chirality. This was used to determine the stereochemistry of chiral centers at C7, C8, and C10. Because C7 and C8 are adjacent stereocenters, these techniques allowed for immediate determination of their relative stereochemistry, however C10 is separated from C8 by C9, which carries two diastereotopic protons. This allows for the determination of relative stereochemistry of C8 and C10 to the C9 protons through [[J-coupling \ 3J coupling]] values, so as to relate the relative stereochemistry of C8 to C10. These methods yielded a relative stereochemistry of 7R, 8S, 10S for the aliphatic chain stereocenters.[6] Stereochemistry at C3 was determined by Marfey's analysis, wherein the compound was ozonized and subsequently hydrolyzed to obtain cysteic acid from the thiazoline ring and attached terminal alkene. Marfey's analysis indicated this amino acid derivative was L-cysteic acid, indicating R absolute stereochemistry at C3.[6] The absolute stereochemistry of the total molecule was determined by synthesizing the possible configurations of the already determined relative chiralities, and comparison of these to natural Kalkitoxin via 13C NMR shift differences, revealing the natural (+)-kalkitoxin stereochemistry to be 3R,7R,8S,10S,2′R.[6]
Structure-activity relationship
The structure-activity relationship (SAR) of a molecule is the connection between the structural moieties within the compound, and how those specific structures directly contribute to the extent and character of the molecule's biological activity. Kalkitoxin exhibits potent cytotoxicity which relies on the complete thiazoline ring for its action.[9] Kalkitoxin analogs lacking the complete thiazoline ring exhibit on the order of 1000-fold decreased toxicity to solid tumor cell lines.[9] This indicates the thiazoline ring structure is a crucial component of kalkitoxin's mechanism of cytotoxicity. The necessity of the stereochemistry exhibited in natural (+)-kalkitoxin decreases moving towards the chiral centers in the core of the molecule, while the more terminal chiral centers and amide methyl group are increasingly crucial for toxicity.[7] In a study which assayed for the toxicity of kalkitoxin and various analogs against brine shrimp, the analogs which experienced the least significant loss of potency were epimers at either C8 or C10.[7] This indicates that C8 and C10 chiralities in natural (+)-kalkitoxin are the least critical for toxic biological activity. It is apparent that C10 chirality is less critical than C8, because the epimer of (+)-kalkitoxin at C10 is more potent than the epimer at C8.[7] Furthermore, the removal of the C10 methyl group has a smaller impact on potency than does epimerization of C7, supporting the trend of decreased SAR correlation at core chiral centers on the aliphatic chain.[7] Epimerization at C3, the attachment point of the terminal alkene to the thiazoline ring, further decreases potency of kalkitoxin, in agreement with the thiazoline ring and overall conformation of the leftmost segment of the molecule being critical for bioactivity. Finally, replacement of the tertiary amide with a secondary amide eliminates any observable toxicity, so this structure is crucial in the mechanism of kalkitoxin toxicity.[7]
Synthesis
Wu et al. synthesis
This effort was the first total synthesis of (+)-kalkitoxin, and served the purpose of deducing the specific stereochemistry of natural kalkitoxin.[6] This synthesis began from an alcohol bearing the proper chirality at C8 and C10 found in (+)-kalkitoxin, and carried a dimethylphenylsiloxy (DPSO) group positioned beta to C8, and a terminal alkene positioned alpha to C10. Hydroboration of this alkene gives the resulting alcohol, which is converted to an azide, which is the position at which (R)-2-methylbutyric acid is coupled to produce the sec-butyl group and amide group. The amide is subsequently methylated, finalizing the tertiary amide which has been shown to be so crucial for kalkitoxin toxicity.[7] O-Desilylation and oxidation of the resulting alcohol produce an acceptor for a Horner-Wadsworth-Emmons reaction, wherein a carbonyl and an alpha-methylated phosphonate react to produce an alkene. In this case, a beta-keto phosphonate bearing an (R)-phenylglycine-derived auxiliary group was ligated to the molecule. This group is lost in asymmetric conjugate addition of an (R)-amino alcohol, which, through two cyclodehydration steps using Wipf's oxazoline-thiazoline interconversion protocol, produces the thiazoline ring.[6]
White et al. synthesis
The second total synthesis of (+)-kalkitoxin was only 16 steps and gave a 3% overall yield.[10] A major aspect by which this differs from the first total synthesis of (+)-kalkitoxin is that rather than using a Horner-Wadsworth-Emmons reaction to ligate a phosphonate carrying the 4-phenyl-2-oxazolidinone, an organocopper conjugate addition reaction was used instead.[10] This was done specifically by connecting the organocopper species to a 4-phenyl-2-oxazolidinone carrying an (S)-N-trans-crotonyl group through a 1,4-nucleophilic addition to the α,β-unsaturation of the crotonyl group. This method is advantageous because it allows for stereoselectivity of the resulting 1,3-dimethyl configuration during the larger sequential introduction of the methyl substituents at the C7, C8 and C10 chiral centers.
Another point of diversion between these two syntheses is the number of carbons separating the keto-auxiliary group from the chiral center at C7. This group was separated by one carbon from C7 in the first total synthesis, so the keto-auxiliary moiety could be converted to a carboxylic acid, in anticipation of addition of the amino alcohol immediately thereafter.[6] In this synthesis, this keto-auxiliary group is directly adjacent to C7, necessitating a one carbon homologation, before construction of the thiazoline ring. This was achieved through reductive bond cleavage of the auxiliary group to a primary alcohol and oxidation to the corresponding aldehyde, Wittig reaction using an ylide carrying a methoxy group to produce an enol ether, hydrolysis to the aldehyde and finally oxidation to produce the carboxylic acid.[10]
Balieu et al. synthesis
This synthesis differentiates itself in that it takes an "assembly line" synthesis approach, as opposed to the conventional iterative synthetic approach taken in previous syntheses which normally necessitate functional-group interconversions and repetitive purifications for aliphatic chain extensions, such as the one found in kalkitoxin. This novel approach is achieved through the use of reagent-controlled chain extension of a boronic ester,[11] which relies on a spontaneous 1,2-migration after formation of an intermediate compound incorporating a newly added lithiated benzoate ester building block.[12]
This allows for control of chirality at each addition by selecting the chirality of each benzoate ester added. Furthermore, this avoids repetitive interconversion and purification steps normally required for repeat chain extensions, which increases yield and efficiency and decreases labor. This synthesis capitalized on this technique by producing the core aliphatic chain as a single large fragment, and coupled this fragment to the chiral sec-butyl group bearing a carboxylic acid.[12] The opposing amino thioether fragment was synthesized separately, and then adjoined and subsequently cyclized following the procedure devised by White et al.[10] In total, this synthesis requires only 7 steps if the initial homologation series is counted as one step.[12]
Targets
Kalkitoxin may activate the NMDA receptor.[1] It also blocks the voltage-gated sodium channel[13] and the electron transport chain (ETC) complex 1.[13] It remains unknown how exactly kalkitoxin binds to the voltage-gated sodium channel. Neurotoxin sit 1 and 2 have been ruled out as possible binding sites, whereas neurotoxin site 7 is suggested as binding site for kalkitoxin.[4] This is probable, because there is inhibition of the channel by kalkitoxin when deltamethrin, which has positive allosteric effects, is present.[13] This could be because molecular determinants for binding are similar in kalkitoxin and deltamethrin.[13]
Mode of action
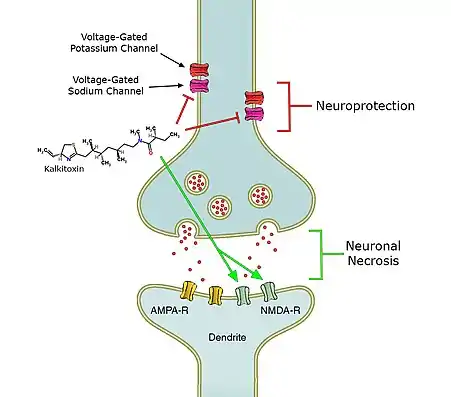
Kalkitoxin induces delayed neuronal necrosis in cerebellar granule cells of the rat.[1] This neuronal necrosis proved to be NMDA-receptor mediated.[1] These receptors are normally activated by glutamate and other excitotoxic compounds and can induce neuronal necrosis.[1] It is not yet known if the toxin induces necrosis directly or via the release of excitotoxic compounds.[1]
Secondly, kalkitoxin blocks voltage-gated sodium channels, thereby inhibiting Ca2+ release that normally occurs when the voltage-gated sodium channel is activated, in a concentration dependent matter.[13] Calcium release has been shown to induce lactate dehydrogenase (LDH) production.[13] The amount of LDH is a measure for neuronal cell death.[13] In the presence of kalkitoxin there is also a concentration-dependent inhibition of neuronal cell death and LDH production (9). The mechanism behind this inhibition is still unknown.[13]
Thirdly, kalkitoxin blocks the electron transport chain (ETC) complex 1,[2] one of the protein complexes involved in mitochondrial respiration.[2] By blocking the ETC complex 1, kalkitoxin potently inhibits hypoxia-inducible factor-1 (HIF-1) activation.[2] HIF-1 is a transcription factor, which enhances the expression of genes that increase oxygen availability, as well as genes that decrease oxygen consumption.[2] Inhibition of HIF-1, which is one of the main effects of kalkitoxin, thus induces cellular hypoxia.
Toxicity
Kalkitoxin is ichthyotoxic to goldfish (Carassius auratus, LC50: 700nM) and to aquatic crustacean brine shrimp (Artemia salina, LC50: 150-180nM [7]).[14] Kalkitoxin also has been shown to have delayed neurotoxic effects on cerebellar granule cells of the rat (LC50: 3,86nM).[2]
Therapeutic research
Many efforts to discover cancer therapeutic drugs focus on the screening of novel biomolecules produced and isolated from various plants and animals.[15] These isolated molecules are screened via in-vitro assays to measure their effects in standardized paradigms designed to select for the desired therapeutic effect. Kalkitoxin was originally isolated from Lyngbya majuscula as an effort to collect new molecules for testing as antitumor or antifungal agents.[4] One of the first tests of kalkitoxin tumor-selective cytotoxicity used an in-vitro assay to test solid tumor selectivity of kalkitoxin's previously demonstrated cytotoxicity against the human colon cell line HCT-116.[9] The assay measured the extent of differential cytotoxicity of kalkitoxin and various analogous structures by observing differential cytotoxicity against solid tumor cells, and either non-solid tumor cells such as leukemia cells or normal cells. This test yielded promising results, as kalkitoxin exhibited preferential cytotoxicity for the solid tumor cell test conditions (Colon 38, and HCT-116 cells) as compared to the non-solid tumor and normal cell conditions.[9]
Kalkitoxin exerts this cytotoxic effect through inhibition of the mitochondrial electron transport chain complex 1.[2] This causes the inhibition of hypoxia-induced HIF-1 activation, which is crucial in solid tumor cancers because hypoxia drives tumor angiogenesis, leading to worsening disease stages and increased resistance to treatment. HIF-1 is a transcription factor which induces expression of genes promoting oxygen availability and decreasing oxygen consumption, the effect of which counteracts cellular hypoxia.[16] Therefore, kalkitoxin's HIF-1 inhibitory ability positions it as a potentially promising molecule to counteract the progression of some solid tumor cancers by blocking the tumor proliferative response to hypoxia. The caveat to kalkitoxin's promising anti-proliferative properties is its neurotoxic effects. At concentrations comparable to those required for tumor-selective cytotoxicity, kalkitoxin induces cell death when applied to rat cerebellar granule neurons (CGN) in culture.[2] Kalkitoxin acts as an N-methyl-D-aspartate (NMDA) receptor agonist, and induces cytotoxicity in cultured rat CGNs at delayed time points.[1] Therefore, this effect must be taken into account when considering kalkitoxin or its chemical derivatives for use as a therapeutic option.
References
- Berman; et al. (1999). "Antillatoxin and kalkitoxin, ichthyotoxins from the tropical cyanobacterium Lyngbya majuscula, induce distinct temporal patterns of NMDA receptor-mediated neurotoxicity". Toxicon. 37 (11): 1645–8. doi:10.1016/s0041-0101(99)00108-7. PMID 10482399.
- Morgan; et al. (2015). "Kalkitoxin inhibits angiogenesis, disrupts cellular hypoxic signaling and blocks mitochondrial electron transport in tumor cells". Marine Drugs. 13 (3): 1552–1568. doi:10.3390/md13031552. PMC 4377999. PMID 25803180.
- Nogle and Gerwick (2003). "Diverse Secondary Metabolites from a Puerto Rican Collection of Lyngbya Majuscula". Journal of Natural Products. 66 (2): 217–20. doi:10.1021/np020332c. PMID 12608852.
- Wu, M. (1997). Novel bioactive secondary metabolites from the marine cyanobacterium Lyngbya majuscule, Thesis (M.S.). Oregon State University – via https://www.researchgate.net/publication/33818310_Novel_bioactive_secondary_metabolites_from_the_marine_cyanobacterium_Lyngbya_majuscula.
{{cite book}}: External link in|via= - Royal Society of Chemistry 2015. "ChemSpider Kalkitoxin".
- Wu; et al. (2000). "Structure, Synthesis, and Biological Properties of Kalkitoxin, a Novel Neurotoxin from the Marine Cyanobacterium Lyngbya majuscule". Journal of the American Chemical Society. 122 (48): 12041–12042. doi:10.1021/ja005526y.
- Umezawa; et al. (2011). "Synthesis and Biological Activity of Kalkitoxin and its Analogues". Journal of Organic Chemistry. 77 (1): 357–70. doi:10.1021/jo201951s. PMID 22111947.
- Hawkins, Clifford J.; Lavin, Martin F.; MarshallKaren A., Karen A.; Van den Brenk, Anna L.; Watters, Diane J. (1990-06-01). "Structure-activity relationships of the lissoclinamides: cytotoxic cyclic peptides from the ascidian Lissoclinum patella". Journal of Medicinal Chemistry. 33 (6): 1634–1638. doi:10.1021/jm00168a016. PMID 2342056.
- White, James D.; Xu, Qing; Lee, Chang-Sun; Valeriote, Frederick A (2004). "Total synthesis and biological evaluation of (+)-kalkitoxin, a cytotoxic metabolite of the cyanobacterium Lyngbya majuscula". Organic & Biomolecular Chemistry. 2 (14): 2092–2102. doi:10.1039/B404205K. PMID 15254638.
- White, James D; Lee, Chang-Sun; Xu, Qing (2003). "Total synthesis of (+)-kalkitoxin". Chemical Communications (16): 2012–2013. doi:10.1039/B306124H. PMID 12934887.
- Burns; et al. (2014). "High Precision Assembly Line Synthesis for Molecules with Tailored Shapes". Nature. 513 (7517): 183–188. doi:10.1038/nature13711. PMC 4167605. PMID 25209797.
- Balieu; et al. (2015). "Toward Ideality: The Synthesis of (+)-Kalkitoxin and (+)-Hydroxyphthioceranic Acid by Assembly-Line Synthesis". Journal of the American Chemical Society. 137 (13): 4398–4403. doi:10.1021/ja512875g. PMID 25625684.
- LePage; et al. (2005). "The neurotoxic lipopeptide kalkitoxin interacts with voltage-sensitive sodium channels in cerebellar granule neurons". Toxicology Letters. 158 (2): 133–9. doi:10.1016/j.toxlet.2005.03.007. PMID 16039402.
- Sarma, T.A. (2012). Handbook of Cyanobacteria. CRC Press. p. 539. ISBN 9781578088003.
- de Bono; et al. (2003). "The future of cytotoxic therapy: selective cytotoxicity based on biology is the key". Breast Cancer Research. 5 (3): 154–9. doi:10.1186/bcr597. PMC 165009. PMID 12793897.
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71.