Papillary renal cell carcinoma
Papillary renal cell carcinoma (PRCC) is a malignant, heterogeneous tumor originating from renal tubular epithelial cells of the kidney, which comprises approximately 10-15% of all kidney neoplasms.[1] Based on its morphological features, PRCC can be classified into two main subtypes, which are type 1 (basophilic) and type 2 (eosinophilic).[2]
| Papillary renal cell carcinoma | |
|---|---|
 | |
| Papillary renal cell carcinoma, type 1, characterised by tubulopapillary architecture with admixed foamy histiocytes in the papillary cores. | |
| Specialty | Oncology |
| Symptoms | Hematuria, flank pain, palpable abdominal mass |
| Diagnostic method | Medical Image Test (ultrasound, computed tomography, magnetic resonance imaging) |
| Treatment | Nephrectomy, partial nephretocmy, targeted molecular therapy |
| Prognosis | 82-90% for five-year survival rate |
As with other types of renal cell cancer, most cases of PRCC are discovered incidentally without showing specific signs or symptoms of cancer.[3] In advanced stages, hematuria, flank pain, and abdominal mass are the three classic manifestation.[4] While a complete list of the causes of PRCC remains unclear, several risk factors were identified to affect PRCC development, such as genetic mutations, kidney-related disease, environmental and lifestyle risk factors.[5] For pathogenesis, type 1 PRCC is mainly caused by MET gene mutation while type 2 PRCC is associated with several different genetic pathways.[6] For diagnosis, PRCC is detectable through computed tomography (CT) scans or magnetic resonance imaging (MRI), which commonly present a small homogeneous hyposvascular tumor.[7] Nephrectomy or partial nephrectomy is usually recommended for PRCC treatment, often accompanied with several targeted molecular therapies to inhibit metastatic spread.[8][9] PRCC patients are predominantly male with a mean age of 52–66 years.[10] When compared to conventional clear cell renal cell carcinoma (RCC), the prognosis of non-metastatic PRCC is more favorable, whereas a relatively worse outcome was reported in patients with metastatic disease.[11] Globally, the incidence of PRCC ranges between 3,500 to 5,000 cases, while it greatly varies depending on gender, age, and race/ethnicity.[12]
Classification
In 2014, PRCC was first acknowledged as a renal tumor subtype by the World Health Organization (WHO) considering its distinct genetic, molecular and histologic characteristics.[10] It is further divided into type 1 and type 2 based on morphological features.[13]
Type 1 Papillary Renal Cell Carcinoma
Type 1 PRCC, also known as a renal tumor caused by a genetic predisposition of hereditary papillary renal cancer syndrome, compromises approximately 25% of all PRCCs.[14][15] In the perspective of immunochemistry, it has a profile of strong CK7 and alpha-methyl acyl-CoA racemase (AMACR) expression at most focal CA-IX expression.[16] Histologically, its epithelium is composed of relatively small-sized simple cuboidal cells lined in a single layer.[17] These cells are well-characterized by basophilic cytoplasm.[18] Due to its solid growth, an extremely compact papillary architecture is often observed.[16] Other morphological characteristics include intracellular hemosiderin and foamy macrophages placed inside of papillary fibrovascular cores or psammoma bodies.[19] In general, the nuclei of type 1 PRCC belong to grade 1-2 of the Fuhrman system.[16]
 A micrograph of type 1 PRCC, illustrating features of small basophilic cells with scarce cytoplasm. A single layer of cells are surrounding the basal membrane.[20] Foamy macrophages are inside of papillary fibrovascular cores.
A micrograph of type 1 PRCC, illustrating features of small basophilic cells with scarce cytoplasm. A single layer of cells are surrounding the basal membrane.[20] Foamy macrophages are inside of papillary fibrovascular cores. Type 1 PRCC, this case being more compact at right.
Type 1 PRCC, this case being more compact at right.
Type 2 Papillary renal cell carcinoma

Accounting for 25% of PRCCs, type 2 PRCC is the pathological subtype that is most commonly associated with hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome.[14][15] When compared to type 1, it shows more variation in protein expression mostly by loss of CK7.[16] In a gross examination, it shows papillae covered by large cells abundant in eosinophilic cytoplasm.[18] Its large spherical nuclei on papillary cores are arranged in a pseudo-stratified manner.[16] Unlike type 1 PRCC, foamy macrophages and psammoma bodies are less common in case of type 2.[15] The majority of type 2 PRCC has high Fuhrman grade nuclei with prominent nucleoli.[16]
Signs and Symptoms
Due to its asymptomatic nature, PRCC is often undetectable, and the majority of cases are incidentally diagnosed during the radiological workup of unrelated diseases.[3][8] Its clinical manifestations are similar to those of clear cell RCC, which are the classical triad of renal cell carcinoma (hematuria, flank pain and palpable abdominal mass; only 6-10% of patients) or even nonspecific symptoms including fatigue, weight loss, fever, and anorexia.[4][10][21] Since early diagnosis is relatively uncommon, PRCC patients may experience symptoms caused by the metastatic spread to secondary sites. Specifically, metastasis occurs most frequently in the lungs followed by bone and the brain, exhibiting a wide range of symptoms including bone pain to a persistent cough.[8][21]
Causes
Currently, the exact cause of PRCC remains unclear. Possible risk factors have been identified that contribute to PRCC development, which include genetic mutations, hereditary syndrome, renal injuries, and lifestyle factors. Germline mutation of c-MET oncogene and fumarate hydratase gene elevates the risk of type 1 and type 2 PRCC respectively through distinct signaling pathways.[22][23] Regarding hereditary conditions, patients with hereditary papillary renal cancer syndrome showed a greater risk of type 1 PRCC, whereas those with hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome have an increased risk of type 2 PRCC.[24][5] Moreover, patients who experienced chronic kidney diseases or acute kidney injury exhibited a higher incidence of PRCC.[25][26] Additionally, other risk factors such as smoking, obesity, and high blood pressure can influence the pathogenesis of PRCC.[5]
Pathogenesis
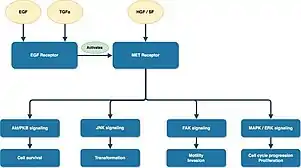
Different molecular mechanisms are involved in PRCC development, which further result in distinct histologic features and clinical outcomes.
Type 1 PRCC is caused by a genetic mutation or a gain in chromosome 7 where the MET gene is positioned, resulting in the promotion of oncogenic pathways in renal epithelial cells.[6] Typically, the MET gene is upregulated for renal tissue repair and regeneration by encoding the receptor tyrosine kinase c-MET of hepatocyte growth factor.[6] However, activation of the oncogenic pathway in the MET gene will manifest invasion, anti-apoptosis, angiogenesis, and metastasis.[6]
Type 2 PRCC is associated with irregularity of several signaling pathways, which includes CDKN2A silencing, mutation in chromatin-modifying genes, and a GpG island methylator phenotype (CIMP).[27] CDKN2A is a tumor suppressor gene, while loss of its expression results in enhanced tumorigenesis and metastasis.[27][28] Moreover, mutation of gene involved in chromatin remodeling (SETD2, BAP1, or PBRM) may lead to higher rate of TFE3/TFEB fusion.[27] Additionally, CIMP papillary renal cell carcinoma tumors exhibited somatic FH gene mutation, which is closely associated with HLRCC syndrome.[23]
Diagnosis
Currently, cross-sectional imaging with computed tomography (CT) and magnetic resonance imaging (MRI) is known as the best option for diagnosing papillary renal tumors.[29]

Computed tomography (CT)
Contrast-enhanced computed tomography (CT) is most commonly used to identify the subtypes of RCC. PRCC can be differentiated from other types of RCC due to its distinguishing features, displaying a small hypovascular renal tumor on T2 weighted images.[7] Typically, PRCC tends to appear homogeneous while clear cell RCC is likely to be in a heterogeneous form when the tumor is less than 3 cm in diameter.[30] Comparatively, in cases of tumors larger than 3 cm in diameter, PRCC is generally heterogeneous with areas of necrosis and hemorrhage compared to chromophobe RCC.[8][31] Solid, small PRCC tumors (<3 cm in diameter) are more easily viewed on nephrographic, excretory phase images rather than on unenhanced, corticomedullary phase images.[8]
Magnetic resonance imaging (MRI)
Magnetic resonance imaging (MRI) is recommended instead of CT for patients with an allergy to iodinated contrast materials.[8] As some renal tumors do not enhance significantly on CT, MRI examination is required to be performed with more sensitive contrast enhancement.[7] On MRI, the distinct features of PRCC are fibrous capsules and homogeneously low single intensity on both T1- and T2-weighted images.[8][32] Specifically, PRCC exhibits hypointensity due to its dense collagenous matrix or deposition of calcium and hemosiderin within the tumor.[33] Such visual features help PRCC to be differentiated from clear cell RCC, which has heterogeneously higher single intensity shown on T2-weighted images.[8][32] PRCC displays the smallest tumor-to-cortex enhancement at corticomedullary and nephrographic phases when juxtaposed with clear cell and chromophobe RCCs.[32]
Grading System


The WHO/ISUP system is histological tumor grading system for renal cell carcinoma, suggested by the International Society of Urologic Pathologists (ISUP) in 2012 to diagnose tumor grades based on nucleolar prominence.[34] Currently recommended by the WHO, this four-tiered WHO/ISUP grading system has also been validated for PRCC.[34]
| Grade | Definition |
|---|---|
| 1 | Nucleoli are absent or inconspicuous and basophilic at 400 magnification. |
| 2 | Nucloeli are conspicuous and eosinophilic at 400 magnification and visible but not prominent at 100 magnification. |
| 3 | Nucleoli are conspicuous and eosinophilic at 100 magnification |
| 4 | There is extreme nuclear pleomorphism, multinucleate giants cells, and/or rhabdoid an/or sarcomatoid differentiation |
Earlier, the Fuhrman system was largely used, and was similarly based on nuclear features.[36]
Treatment
First-line treatment for metastatic PRCC has not been standardized. Thus, similar treatment approaches for clear cell RCC have been used for PRCC, even though it has a distinct tumor histology.[9][37]
Surgery
Nephrectomy or nephron-sharing partial nephrectomy is widely recommended to reduce the risk of metastasis by eliminating all or part of the kidney.[8] Surgery procedures for PRCC depend on the patient's status and are very similar to procedures performed on RCC patients.
Targeted Therapy
Several medications that target molecular pathways in RCC have been possible options for advanced and metastatic PRCC.[9] Among different medications, tyrosine kinase inhibitors (TKIs) and mammalian target of rapamycin (mTOR) inhibitors are effective in inhibiting angiogenesis, blocking growth and suppressing spread of the tumor.[38] Sunitinib, sorafenib, and axitinib are TKIs with anti-vascular endothelial growth factor (VEGF), which inhibit cellular signaling by targeting multiple receptor tyrosine kinase.[38][39][40][41] Everolimus and temsirolimus are used in deregulating the mTOR pathway.[42][43] Specifically, mTOR inhibitors have crucial roles in regulating cell growth, cell proliferation and metabolism of highly active tumor cells.[38] Other targeted agents such as MET inhibitors, epidermal growth factor receptor (EGFR) inhibitors, and monoclonal antibodies, are also promising treatment approaches for PRCC.[9] Foretinib is one example of a multikinase inhibitor targeting c-MET.[44] Considering that MET gene mutation is one oncogenic pathway of PRCC, MET inhibitors like tivantinib and volitinib are currently being investigated as a new targeted therapy option.[9][44]
Prognosis
The five-year survival rate of PRCC has been reported as 82-90%, which is slightly higher than that of other kidney cancers.[45] The reduced survival rate has been positively correlated to several factors, which are high nuclear grade and stage, vascular invasion, DNA aneuploidy, and more.[46] Patients with type 1 PRCC have significantly improved survival rates than those with type 2, which is a reflection of its lower TNM stage with a well-encapsulated tumor.[47] Compared to other common types of RCC, PRCC exhibits a relatively lower risk of tumor recurrence and cancer-related death after nephrectomy.[48] Specifically, the cancer-specific survival rate at five years following surgery with PRCC has reached up to 91%, while clear cell RCC and chromophobe RCC were 72% and 88%, respectively.[46]
Epidemiology
Among different histologic subtypes of RCC, PRCC is the second most predominant type and accounts for 10-15% of all renal tumors.[49] In the case of the United States, it is estimated that the incidence of PRCC will rise to 3,500 to 5,000 cases annually.[12] Generally, PRCC is more prevalent among men than women, while the reported sex ratio (M: F) varies from 1.8:1 to 3.8:1.[50] The mean age at presentation is identified as 52–66 years old; however, no statistically significant difference was found in the incidence of PRCC between the younger (< 40 years) and older adult groups (>40 years).[10][51] In terms of racial variation, several studies have proven that people with African or Afro-Caribbean ancestry tend to have higher chances of being diagnosed with PRCC. According to the National Cancer Database, PRCC was more common in the Non-Hispanic Black population (38.9%) when compared to other races – Asian American (18.0%), Non-Hispanic White (13.2%), and Hispanic White populations (6.1%).[52]
References
- Cairns P (2011). "Renal cell carcinoma". Cancer Biomarkers. 9 (1–6): 461–73. doi:10.3233/CBM-2011-0176. PMC 3308682. PMID 22112490.
- Lee J, Chae HK, Lee W, Nam W, Lim B, Choi SY, et al. (2018-12-28). "Comparison of Prognosis in Types 1 and 2 Papillary Renal Cell Carcinoma and Clear Cell Renal Cell Carcinoma in T1 Stage". The Korean Journal of Urological Oncology. 16 (3): 119–125. doi:10.22465/kjuo.2018.16.3.119.
- Encyclopedia of Cancer. Academic Press. 2018-10-12. ISBN 978-0-12-812485-7.
- Courthod G, Tucci M, Di Maio M, Scagliotti GV (October 2015). "Papillary renal cell carcinoma: A review of the current therapeutic landscape". Critical Reviews in Oncology/Hematology. 96 (1): 100–12. doi:10.1016/j.critrevonc.2015.05.008. hdl:2318/1564896. PMID 26052049.
- "Risk Factors for Kidney Cancer". www.cancer.org. Retrieved 2020-04-08.
- Rhoades Smith KE, Bilen MA (November 2019). "A Review of Papillary Renal Cell Carcinoma and MET Inhibitors". Kidney Cancer. 3 (3): 151–161. doi:10.3233/KCA-190058. PMC 6918905. PMID 31867475.
- Couvidat C, Eiss D, Verkarre V, Merran S, Corréas JM, Méjean A, Hélénon O (November 2014). "Renal papillary carcinoma: CT and MRI features". Diagnostic and Interventional Imaging. 95 (11): 1055–63. doi:10.1016/j.diii.2014.03.013. PMID 25443332.
- Vikram R, Ng CS, Tamboli P, Tannir NM, Jonasch E, Matin SF, et al. (2009-05-01). "Papillary renal cell carcinoma: radiologic-pathologic correlation and spectrum of disease". Radiographics. 29 (3): 741–54, discussion 755–7. doi:10.1148/rg.293085190. PMID 19448113.
- Bourlon MT, Meneses-Medina M, Vázquez-Manjarrez S, Bustamante-Romero FM, Gallegos-Garza AC, Lam ET (November 2015). "A Patient With Newly Diagnosed Metastatic Type 2 Papillary Renal Cell Carcinoma". Oncology (Williston Park, N.Y.). 29 (11): 880–6. PMID 26573066.
- International Agency for Research on Cancer (2004). Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. IARC. ISBN 978-92-832-2415-0.
- Klatte T, Gallagher KM, Afferi L, Volpe A, Kroeger N, Ribback S, et al. (October 2019). "The VENUSS prognostic model to predict disease recurrence following surgery for non-metastatic papillary renal cell carcinoma: development and evaluation using the ASSURE prospective clinical trial cohort". BMC Medicine. 17 (1): 182. doi:10.1186/s12916-019-1419-1. PMC 6775651. PMID 31578141.
- Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, et al. (2005). "Cancer statistics, 2005". CA: A Cancer Journal for Clinicians. 55 (1): 10–30. doi:10.3322/canjclin.55.1.10. PMID 15661684. S2CID 22356919.
- Weidner N, Cote RJ, Suster S, Weiss LM (2009-07-08). Modern Surgical Pathology E-Book. Elsevier Health Sciences. ISBN 978-1-4377-1958-1.
- Haas NB, Nathanson KL (January 2014). "Hereditary kidney cancer syndromes". Advances in Chronic Kidney Disease. 21 (1): 81–90. doi:10.1053/j.ackd.2013.10.001. PMC 3872053. PMID 24359990.
- "Papillary renal cell carcinoma (PRCC)". www.pathologyoutlines.com. Retrieved 2020-04-08.
- Warren AY, Harrison D (December 2018). "WHO/ISUP classification, grading and pathological staging of renal cell carcinoma: standards and controversies". World Journal of Urology. 36 (12): 1913–1926. doi:10.1007/s00345-018-2447-8. PMC 6280811. PMID 30123932.
- Chevarie-Davis M, Riazalhosseini Y, Arseneault M, Aprikian A, Kassouf W, Tanguay S, et al. (July 2014). "The morphologic and immunohistochemical spectrum of papillary renal cell carcinoma: study including 132 cases with pure type 1 and type 2 morphology as well as tumors with overlapping features". The American Journal of Surgical Pathology. 38 (7): 887–94. doi:10.1097/PAS.0000000000000247. PMID 24919183. S2CID 1998988.
- Bukowski RM, Novick A (2009-12-03). Clinical Management of Renal Tumors. Springer Science & Business Media. ISBN 978-1-60327-149-3.
- "Right Kidney". College of American Pathologists. Retrieved 2020-04-19.
- Muglia VF, Prando A (2014-04-10). "Renal cell carcinoma: histological classification and correlation with imaging findings". Radiologia Brasileira. 48 (3): 166–74. doi:10.1590/0100-3984.2013.1927. PMC 4492569. PMID 26185343.
- "papillary renal cell carcinoma". www.ebi.ac.uk. Retrieved 2020-04-08.
- "What Causes Kidney Cancer?". www.cancer.org. Retrieved 2020-04-08.
- Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, Davis C, et al. (January 2016). "Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma". The New England Journal of Medicine. 374 (2): 135–45. doi:10.1056/NEJMoa1505917. PMC 4775252. PMID 26536169.
- "Hereditary Papillary Renal Carcinoma". Cancer.Net. 2012-06-25. Retrieved 2020-04-08.
- van de Pol JA, van den Brandt PA, Schouten LJ (February 2019). "Kidney stones and the risk of renal cell carcinoma and upper tract urothelial carcinoma: the Netherlands Cohort Study". British Journal of Cancer. 120 (3): 368–374. doi:10.1038/s41416-018-0356-7. PMC 6353869. PMID 30563989.
- Peired AJ, Antonelli G, Angelotti ML, Allinovi M, Guzzi F, Sisti A, et al. (March 2020). "Acute kidney injury promotes development of papillary renal cell adenoma and carcinoma from renal progenitor cells". Science Translational Medicine. 12 (536): eaaw6003. doi:10.1126/scitranslmed.aaw6003. PMID 32213630. S2CID 214671587.
- Malouf GG, Joseph RW, Shah AY, Tannir NM (May 2017). "Non-clear cell renal cell carcinomas: biological insights and therapeutic challenges and opportunities". Clinical Advances in Hematology & Oncology. 15 (5): 409–418. PMID 28591094.
- Zhao R, Choi BY, Lee MH, Bode AM, Dong Z (June 2016). "Implications of Genetic and Epigenetic Alterations of CDKN2A (p16(INK4a)) in Cancer". EBioMedicine. 8: 30–39. doi:10.1016/j.ebiom.2016.04.017. PMC 4919535. PMID 27428416.
- "Genetics of Kidney Cancer (Renal Cell Cancer) (PDQ®)–Health Professional Version". National Cancer Institute. 2013-11-01. Retrieved 2020-04-08.
- Herts BR, Coll DM, Novick AC, Obuchowski N, Linnell G, Wirth SL, Baker ME (February 2002). "Enhancement characteristics of papillary renal neoplasms revealed on triphasic helical CT of the kidneys". AJR. American Journal of Roentgenology. 178 (2): 367–72. doi:10.2214/ajr.178.2.1780367. PMID 11804895.
- Kim JK, Kim TK, Ahn HJ, Kim CS, Kim KR, Cho KS (June 2002). "Differentiation of subtypes of renal cell carcinoma on helical CT scans". AJR. American Journal of Roentgenology. 178 (6): 1499–506. doi:10.2214/ajr.178.6.1781499. PMID 12034628.
- Gurel S, Narra V, Elsayes KM, Siegel CL, Chen ZE, Brown JJ (2013-03-11). "Subtypes of renal cell carcinoma: MRI and pathological features". Diagnostic and Interventional Radiology. 19 (4): 304–11. doi:10.5152/dir.2013.147. PMID 23439256.
- Hiraki Y, Okamoto D, Nishie A, Asayama Y, Ishigami K, Ushijima Y, et al. (August 2019). "Papillary renal cell carcinoma with massive hematoma mimicking hemangioma". Radiology Case Reports. 14 (8): 1003–1006. doi:10.1016/j.radcr.2019.05.025. PMC 6556879. PMID 31198484.
- Moch H (July 2016). "[The WHO/ISUP grading system for renal carcinoma]". Der Pathologe. 37 (4): 355–60. doi:10.1007/s00292-016-0171-y. PMID 27271258. S2CID 8644427.
- Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM (July 2016). "The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours". European Urology. 70 (1): 93–105. doi:10.1016/j.eururo.2016.02.029. PMID 26935559.
- Table 3 from: Weiss, Lawrence M.; Gelb, Arnold B.; Jeffrey Medeiros, L. (1995). "Adult Renal Epithelial Neoplasms". American Journal of Clinical Pathology. 103 (5): 624–635. doi:10.1093/ajcp/103.5.624. ISSN 0002-9173.
- Liu K, Ren Y, Pang L, Qi Y, Jia W, Tao L, et al. (2015-07-01). "Papillary renal cell carcinoma: a clinicopathological and whole-genome exon sequencing study". International Journal of Clinical and Experimental Pathology. 8 (7): 8311–35. PMC 4555730. PMID 26339402.
- Zarrabi K, Fang C, Wu S (February 2017). "New treatment options for metastatic renal cell carcinoma with prior anti-angiogenesis therapy". Journal of Hematology & Oncology. 10 (1): 38. doi:10.1186/s13045-016-0374-y. PMC 5288948. PMID 28153029.
- Ravaud, A.; Oudard, S.; De Fromont, M.; Chevreau, C.; Gravis, G.; Zanetta, S.; Theodore, C.; Jimenez, M.; Sevin, E.; Laguerre, B.; Rolland, F.; Ouali, M.; Culine, S.; Escudier, B. (2015-06-01). "First-line treatment with sunitinib for type 1 and type 2 locally advanced or metastatic papillary renal cell carcinoma: a phase II study (SUPAP) by the French Genitourinary Group (GETUG)†". Annals of Oncology. 26 (6): 1123–1128. doi:10.1093/annonc/mdv149. PMID 32018840.
- Stadler WM, Figlin RA, McDermott DF, Dutcher JP, Knox JJ, Miller WH, et al. (March 2010). "Safety and efficacy results of the advanced renal cell carcinoma sorafenib expanded access program in North America". Cancer. 116 (5): 1272–80. doi:10.1002/cncr.24864. PMID 20082451. S2CID 205659715.
- Kamai T, Abe H, Arai K, Murakami S, Sakamoto S, Kaji Y, Yoshida KI (March 2016). "Radical nephrectomy and regional lymph node dissection for locally advanced type 2 papillary renal cell carcinoma in an at-risk individual from a family with hereditary leiomyomatosis and renal cell cancer: a case report". BMC Cancer. 16: 232. doi:10.1186/s12885-016-2272-7. PMC 4794818. PMID 26983443.
- Zanardi E, Verzoni E, Grassi P, Necchi A, Giannatempo P, Raggi D, et al. (June 2015). "Clinical experience with temsirolimus in the treatment of advanced renal cell carcinoma". Therapeutic Advances in Urology. 7 (3): 152–61. doi:10.1177/1756287215574457. PMC 4485412. PMID 26161146.
- Coppin C (May 2010). "Everolimus: the first approved product for patients with advanced renal cell cancer after sunitinib and/or sorafenib". Biologics: Targets and Therapy. 4: 91–101. doi:10.2147/btt.s6748. PMC 2880340. PMID 20531964.
- Choueiri TK, Vaishampayan U, Rosenberg JE, Logan TF, Harzstark AL, Bukowski RM, et al. (January 2013). "Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma". Journal of Clinical Oncology. 31 (2): 181–6. doi:10.1200/JCO.2012.43.3383. PMC 3532390. PMID 23213094.
- Nosé V (2020-02-14). Diagnostic Pathology: Familial Cancer Syndromes E-Book. Elsevier Health Sciences. ISBN 978-0-323-71205-7.
- Bostwick DG, Cheng L (2008-01-01). Urologic Surgical Pathology. Elsevier Health Sciences. ISBN 978-0-323-01970-5.
- "MDEdge Oauth". profreg.medscape.com. Retrieved 2020-04-08.
- Zucchi A, Novara G, Costantini E, Antonelli A, Carini M, Carmignani G, et al. (April 2012). "Prognostic factors in a large multi-institutional series of papillary renal cell carcinoma". BJU International. 109 (8): 1140–6. doi:10.1111/j.1464-410X.2011.10517.x. PMID 21871053. S2CID 205545457.
- Singh SR, Rameshwar P (2014-04-21). MicroRNA in Development and in the Progression of Cancer. Springer Science & Business. ISBN 978-1-4899-8065-6.
- Fernandes DS, Lopes JM (2015-01-01). "Pathology, therapy and prognosis of papillary renal carcinoma". Future Oncology. 11 (1): 121–32. doi:10.2217/fon.14.133. PMID 25572787.
- Suh JH, Oak T, Ro JY, Truong LD, Ayala AG, Shen SS (2009-01-30). "Clinicopathologic features of renal cell carcinoma in young adults: a comparison study with renal cell carcinoma in older patients". International Journal of Clinical and Experimental Pathology. 2 (5): 489–93. PMC 2655150. PMID 19294008.
- Batai K, Harb-De la Rosa A, Zeng J, Chipollini JJ, Gachupin FC, Lee BR (November 2019). "Racial/ethnic disparities in renal cell carcinoma: Increased risk of early-onset and variation in histologic subtypes". Cancer Medicine. 8 (15): 6780–6788. doi:10.1002/cam4.2552. PMC 6826053. PMID 31509346.