Renal–hepatic–pancreatic dysplasia
Renal–hepatic–pancreatic dysplasia is an autosomal recessive congenital disorder characterized by pancreatic fibrosis, renal dysplasia and hepatic dysgenesis. It is usually fatal soon after birth.An association with NPHP3 has been described.[1] It was characterized in 1959.[2][3]
| Renal–hepatic–pancreatic dysplasia | |
|---|---|
| Other names | Ivemark II syndrome, Renohepaticopancreatic dysplasia |
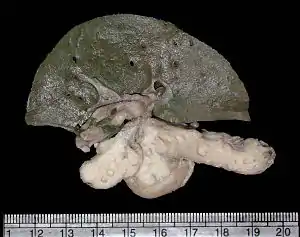 | |
| Gross photo of liver and pancreas showing multiple cysts in the latter in a patient with renal–hepatic–pancreatic dysplasia | |
References
- Bergmann C, Fliegauf M, Brüchle NO, et al. (April 2008). "Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia". Am. J. Hum. Genet. 82 (4): 959–970. doi:10.1016/j.ajhg.2008.02.017. PMC 2427297. PMID 18371931.
- Vankalakunti M, Gupta K, Kakkar N, Das A (2007). "Renal-hepatic-pancreatic dysplasia syndrome (Ivemark's syndrome)". Diagn Pathol. 2: 24. doi:10.1186/1746-1596-2-24. PMC 1919354. PMID 17605805.
- IVEMARK BI, OLDFELT V, ZETTERSTROM R (January 1959). "Familial dysplasia of kidneys, liver and pancreas: a probably genetically determined syndrome". Acta Paediatr. 48 (1): 1–11. doi:10.1111/j.1651-2227.1959.tb16011.x. PMID 13626573. S2CID 196353197.
External links
This article is issued from Wikipedia. The text is licensed under Creative Commons - Attribution - Sharealike. Additional terms may apply for the media files.