Richter's transformation
Richter's transformation (RT), also known as Richter's syndrome, is the conversion of chronic lymphocytic leukemia (CLL) or its variant, small lymphocytic lymphoma (SLL), into a new and more aggressively malignant disease.[1] CLL is the circulation of malignant B lymphocytes with or without the infiltration of these cells into lymphatic or other tissues while SLL is the infiltration of these malignant B lymphocytes into lymphatic and/or other tissues with little or no circulation of these cells in the blood.[2] CLL along with its SLL variant are grouped together in the term CLL/SLL.[1]
| Richter's transformation | |
|---|---|
| Other names | Richter's syndrome |
| Specialty | Hematopathology, Oncology, Surgical oncology |
| Types | Diffuse large B-cell lymphoma type, Hodgkin's lymphoma type |
| Prognosis | Poor |
| Frequency | rare |
RT is diagnosed in individuals who have CLL/SLL that converts to a malignancy with the microscopic histopathology of diffuse large B-cell lymphoma (DLBCL) or, less commonly, Hodgkin’s lymphoma (HL).[3] There are rare cases of: 1) CLL/SLLs that convert into lymphoblastic lymphoma, hairy cell leukemia, or a high grade T cell lymphoma[4] such as anaplastic large-cell lymphoma or angioimmunoblastic T-cell lymphoma;[5] 2) CLL/SLLs that convert into acute myeloid leukemia;[6] 3) CLL/SLLs that convert into non-hematological malignancies such as lung cancer, brain cancer, melanoma of the eye or skin,[5][7] salivary gland tumors, and Kaposi's sarcomas;[8] and 4) conversion of follicular lymphoma, lymphoblastic lymphoma, or marginal zone lymphoma into other types of hematological malignancies.[9] While some of these conversions have been termed RTs, the World Health Organization[10] and most reviews have defined RT as a conversion of CLL/SLL into a disease with DLBCL or HL histopathology. Here, RTs are considered to be CLL/SLLs which convert into a disease with either DLBCL histopathology (here termed DLBCL-RT) or Hodgkin's lymphoma histopathology (here termed HL-RT).
CLL/SLL is the most common adult leukemia in Western countries, accounting for 1.2% of the new cancers diagnosed each year in the United States. It usually occurs in older adults (median age at diagnosis 70) and follows an indolent course over many years.[11] About 1-10% of CLL/SLLs develop a Richter's transformation at a rate of 0.5–1% per year. In earlier studies, the transformed disease was reported to be far more aggressive than CLL/SLL with overall median survival times (i.e. times in which 50% of cases remain alive) between 1.1 and 16.3 months. Newer therapeutic regimens are improving the prognosis of DLBCL-RT and HL-RT.[12]
History
In 1928 Maurice Richter reported that a patient with CLL developed an aggressive generalized swelling of his lymph nodes, liver, and spleen due to their infiltration by rapidly growing "sarcoma-like cells". The patient died of this disease 22 days after his presentation. Dr. Richter termed the disorder "generalized reticular cell sarcoma".[13] In 1964, Lortholary et al. described the occurrence of DLBCL in CLL patients and named the condition Richter's transformation.[14] Subsequent studies have combined SLL with CLL and included HL-RT with DLBCL-RT in the definition of CLL/SLL RTs.[4]
Presentation
Studies have reported that CLL/SLL transforms into DLBCL-RT in ~90% and into HL-RT in 0.7-15% of all RTs.[1] These transformations can occur at any point in the course of CLL/SLL. In a study of 77 individuals, DLBCL-RT and HL-RT were diagnosed simultaneously with CLL/SLL in 6 cases or 3-171 months after being diagnosed with CLL/SLL in 71 cases.[15] A study of 10 RT cases reported that one individual presented with transformed CLL/SLL and 9 transformed 12 to 111 months after being diagnosed with CLL/SLL.[9] The median time between the diagnosis of CLL/SLL and RT has varied between 1.8 and 5 years in 5 other studies.[16]
Individuals with CLL/SLL that develop RT typically present with a rapid increase in the size of their superficial (i.e. cervical, axillary, inguinal, and/or retropharyngeal) lymph nodes; this may be the only sign of the transformation.[2] Other symptoms may include B symptoms (i.e. fever in the absence of infection, drenching night sweats, and/or unexplained weight loss), and/or deterioration in general health. These symptoms are often accompanied by the development of extra-nodal disease, i.e. swelling or tumors due to the infiltration of malignant B lymphocytes into the gastrointestinal tract,[17] bone, skin, central nervous system, spleen, liver,[2] urinary bladder, thyroid gland, and/or pulmonary pleurae.[9] Abnormal laboratory findings include elevation in blood lactate dehydrogenase levels in 50–80% of cases, progressively worsening anemia (i.e. decreases in red blood cells), thrombocytopenia (i.e. decreases in blood platelets),[1] and/or hypercalcemia (i.e. elevation in serum calcium levels often due to bone involvement).[6] FDG-positron emission tomography–computed tomography (i.e. FDG-PET-CT) can determine the sites of tissue invasion, the best sites to biopsy, and in some cases suggest the diagnosis of DLBCL-RT by showing that the involved tissues have distinctively high levels of FDG uptake.[2][12][18] Individuals presenting with RT at the time of CLL/SLL diagnosis will show these symptoms and signs along with microscopic histopathological evidence of CLL/SLL concurrently with DLBCL-RT or HL-RT.
Risk factors for developing Richter's transformation
Individuals with CLL/SLL are considered to be at an increased risk for developing RT if they have: 1) enlarged lymph nodes, liver, and/or spleen; 2) advanced stage disease; 3) low blood platelet counts and/or elevated serum beta-2-microglobulin levels; 4) CLl/SLL cells which develop deletions in the CDKN2A gene, disruptions of the TP53 gene, activation of the C-MYC gene, trisomy (i.e. extra) chromosome 12, or mutations in the NOTCH1 gene;[12] and/or 5) prior CLL/SLL treatment with chemotherapy regimens combining purine analogues and alkylating agents, multiple different types of chemotherapy,[12] and/or combinations of fludarabine, cyclophosphamide, and rituximab (the latter regimen has been associated with a 2.38-fold higher risk of CLL/SLL developing an RT).[8]
Histopathology
DLBCF-RT histopathology
The microscopic histopathology of DLBCL-RT in involved lymph nodes and other tissues stained with hemotoxylin and eosin generally shows confluent sheets of large malignant B lymphocytes that resemble centroblasts in ~80% of cases or immunoblasts in the remaining ~20% of cases. These malignant B lymphocytes express CD20 surface membrane protein in almost all cases, PD-1 surface membrane protein in up to 80% of cases (high PD-1 levels help cancer cells evade host immune systems), CD5 surface membrane protein in ∼30% of cases, and CD23 surface membrane protein in ∼15% of cases. In 90-95% of cases, these cells also express IRF4 (a transcription factor that regulates the development of lymphocytes including B lymphocyte) or in the other 5-10% of cases CD10 (an enzyme found in the neoplastic cells of pre-B cell leukemias and some cases of CLL/SLL).[12]
HL-RT histopathology
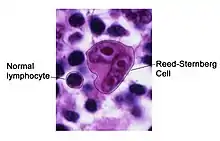
The histopathology of the involved tissues in HL-RT is diagnosed based of the presence of Reed-Sternberg cells (here termed RS cells). The adjacent micrograph shows a typical RS cell, surrounded by normal lymphocytes. RS cells are distinctively large and have multiple nuclei, as in his case, or one nucleus with two lobes. RS cells express CD30 cell surface protein (a member of the tumor necrosis factor receptor family) and CD15 (a blood group antigen carbohydrate on the cell surface).[19] One study reported the RS cells in HL-RT do not express CD20[19] but another reported that ~35% do.[20] These cells also express a protein located in the cell nucleus, lymphoid enhancer-binding factor 1, in ~80% of cases.[19] The RS cells in HL-RT are spread throughout 1) a CLL/SLL-like background of variably shaped, small lymphocytes or 2) an inflammatory cell-like background of epithelioid histiocytes, eosinophils, and plasma cells that is similar to that found in many cases of Hodgkin's lymphoma not due to RT (here termed HL-not RT). HL-RT cases with the CLL/SLL-like background are termed type 1 (or type I) HL-RT and those with the inflammatory cell-like background are termed type 2 (or type II) HL-RT.[1] While some studies have regarded lesions with the type 1 histopathology as not true HL-RTs, one study reported that, among 26 type 1 and 51 type 2 cases, 3 showed an evolution of type 1 into a type 2 histopathology on serial biopsies and the two types were similarly responsive to chemotherapy regimens used to treat Hodgkin's lymphoma-not RT.[20] A second study reported that the type 1 and 2 histopathology can occur not only in succession but also in the same lesion.[21] Finally, a study of 51 type 2 HL-RT cases showed that the RS cells expressed PAX5 in 100%, CD30 in 100%, CD15 in 92%, CD20 in 47%, and ZAP-70 in 32% of case while 26 type 1 cases had RS cells that expressed these respective proteins in 100%, 100%, 78%, 52% , and 57% of the cases.[20] Here, HL-RT is regarded as consisting of leukemic cells with type 1, type 2, or a mixture of the type 1 with type 2 histopathology.
Epstein-Barr virus in RT
More than 90% of the world's population is infected with the Epstein-Barr virus (EBV). During the infection, EBV enters B lymphocytes and may cause infectious mononucleosis, minor non-specific symptoms, or no symptoms. The virus then goes into a latency phase in which infected individuals become lifetime asymptomatic carriers of EBV in their B lymphocytes. Long after its initial infection and latency, EBV may again become active in the B lymphocytes and cause a wide range of EBV-associated diseases including various Epstein–Barr virus–associated lymphoproliferative diseases.[22] EBV reactivation can occur in the B lymphocytes of CLL/SLL and lead to a severer form of CLL/SLL and/or to Richter's transformation.[23] RT transformation has been reported to rarely underlie the development of DLBCL-RT but is associated with the development of type 1 HL-RT in 75% of 51 type 2 cases and 65% of 26 type 1 cases.[20] EBV is detected in 30% to 50% of HL-not RT cases (see Epstein–Barr virus-positive Hodgkin lymphoma).[22] EBV infection in CLL/SLL malignant B lymphocytes is often diagnosed using In situ hybridization to detect Epstein–Barr virus–encoded small RNAs (i.e. EBERs) made by the virus.[19]
Gene abnormalities
DLBCL-RT gene abnormalities
The malignant B lymphocytes in DLBCL-RT but not DLBCL unrelated to RT (i.e. DLBCL-not RT) carry an inactivated TP53 tumor suppressor gene in 50–60% of cases[12] and often abnormalities in their NOTCH1, MYC, and CDKN2A genes.[1] DLBCL-RT cells usually do not have abnormalities in the genes regulating cell signaling or B lymphocyte differentiation pathways that are often found in the malignant B lymphocytes of DLBCLs-not RT. DLBCL-RT malignant B lymphocytes also lack gene abnormalities commonly found in DLBCLs-not RT such as inactivated acetyltransferase, CREB-binding protein, EP300, beta-2 microglobulin genes; translocations of the BCL6 and BCL2 genes; and losses of PRDM1 and TNFAIP3 genes.[1]
There is an important distinction in DLBCL-RTs based on the similarities of their antibody-producing genes to those in their preceding CLL/SLL's malignant B lymphocytes. Normal B lymphocytes make antibodies that recognize and bind to foreign antigens. The formation of these antibodies requires the rearrangement of antibody-producing genes (see antibody production). Analyses indicated that ~80% of DLBCL-RT cases have antibody-producing genes in their malignant B lymphocytes that are related to the antibody-producing genes in their precedent CLL/SLL's malignant B lymphocytes; these cases represent true transformations of CLL/SLL malignant B lymphocytes and are here termed pure DLBCL-RTs. The remaining ~20% of DLBCL cases do not have such a relationship and therefore do not originate from their predecessor CLL/SLL's malignant B lymphocytes.[1][3] These cases are here termed de novo DLBCL-RTs.[3] A Surveillance, Epidemiology, and End Results review of 530 cases diagnosed with RT reported that pure DLCBL-RT and de novo DLBCL-RT cases had median survival times of 14.2 and 62.5 months, respectively.[17] Two smaller studies reported that pure DLBCLs-RT and de novo DLBCL-RT cases had median survival times of 8-16 months and ~ 60 months, respectively,[24] and 14.2 and 62.5 months, respectively.[1] Thus, pure DLBCL-RT is a far more aggressive disease than de novo DLBCL-RT.
HL-RT gene abnormalities
The RS cells in HL-RT may also show antibody-producing gene rearrangements that differ from those in their preceding CLL/SLL cells.[1][19] One study found that 53% of 14 type 2 HL-RT cases had, and 47% did not have, antibody-producing gene changes in their RS cells that were related to those in their predecessor CLL/SLL malignant B lymphocytes while 29% of 14 type 1 HL-RT cases had, and 71% did not have, antibody-producing genes that were related to their preceding CLL/SLL B lymphocytes.[20] Thus, HL-RT cases, like DLBC-RTL cases, may be either evolutions of the disease from their CLL/SLL malignant B lymphocytes or not have this relationship.[12] Notably, Type 1 DL-RT is a more aggressive disease than type 2 HL-RT (see THL-RT treatment and prognosis section).[21]
Diagnosis
The diagnosis of RT depends on finding that individuals with a history of stable CLL/SLL or who present for the first time with CLL/SLL have: 1) rapidly worsening symptoms and/or signs, particularly enlarging lymph nodes or lesions in non-lymph node tissues (see Presentation section);[2] 2) FDG-PET-CT scans that may show involved tissues have high levels of FDG uptake;[18] 3) excisional biopsy (surgical removal of an involved tissue such as a lymph node) or core biopsy (surgical removal of a portion of an involved tissue) which shows the histopathology of DLBCL-RT or HL-RT (Fine-needle aspiration biopsies of involved tissues have not been clinically useful, accurate, or valuable in diagnosing CLL/SLL-RT.[1]); and 4) gene and/or protein expression analyses that can differentiate pure DLBLC-RT from de novo DLBCL-RT (see DLBCL-RT gene abnormalities section).[1] A subgroup of CLL/SLL (∼23% of all cases) develop "accelerated" CLL, i.e. malignant B lymphocytes that are proliferating rapidity.[11] Individuals with accelerated CCL/SLL show worsening symptoms and signs as well as a microscopic histopathology of their involved tissues that can be difficult to distinguish from RTs.[10] FDG-PET-CT scans may help distinguish RT from accelerated CLL/SLL if they show that the involved tissue in RT take-up very high levels of FDG.[2][18] Nonetheless, the diagnosis of these RTs can be difficult to distinguish form each other as well as from accelerated CLL: the final diagnosis of RTs should be made by a hematopathologist familiar with this area.[12]
Treatment and prognosis
DLBCL-RT treatment and prognosis
As of 2021, there were no published randomized controlled trials that defined the optimal treatment for RT. DLBCL-RT cases have been treated with chemotherapy (therapy targeting the cancer cells) combined with immunotherapy (therapy targeting the immune system). The modified CHOP chemoimmunotherapy regimen termed [R]-CHOEP, which consists of rituximab (an immunotherapy antibody preparation that binds to CD20 cell surface protein), cyclophosphamide, doxorubicin, vincristine, and prednisone, has given overall response rates of 50–60% with median overall survival times of 15–21 months. Other chemoimmunotherapy regimens have been used to treat DLBCL-RT. The R-EPOCH regimen of rituximab, etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin gave an overall response rate of 39% and a median overall survival rate of 5.9 months in a retrospective cohort study. R-DHAP (rituximab, dexamethasone, cytarabine, and cisplatin), R-ESHAP (rituximab, etoposide, methylprednisolone, cytarabine, and cisplatin), and dose-intensified regimens such as R-hyper-CVAD (rituximab with hyper-fractionated [i.e. intensive treatment with small doses given more than once per day] cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with high-dose methotrexate and cytarabine as described elsewhere[25]) have given higher complete response rates but also higher adverse events, higher treatment-related mortality, and shorter overall survival times compared to [R]-CHOEP. Consequently, these other regimens are not commonly used to treat DLBCL-RT. Recently, small molecule anti-cancer drugs (e.g. venetoclax, atezolizumab, duvelisib, ublituximab, zanubrutinib, obinutuzumab, olatuzumab, and blinatumomab) which are used to treat CLL/SLL and/or DLBCL-not RT have been added to chemotherapy and immunotherapy regimens to treat RT. While these studies are still in their early stages, i.e. stages I or II, some have produced encouraging results.[1] Further studies of these small molecule drugs as well as studies using CAR-T technology to treat DLBCL-RT are underway.[12] Chemotherapy combined with immunotherapy is and will remain the gold standard for treating DLBCL-RT until future studies report on more effective regimens.[12]
Patients with DLBCL-RT have been treated with autologous or allogenic hematopoietic stem cell transplantation. In these procedures, hematopoietic stem cells are isolated from the patient for an autologous or from a donor for allogenic transplant. The patients are then treated with an "ablation therapy regimen", i.e. high-dose chemotherapy with or without immunotherapy and radiotherapy to eradicate or at least stop or reduce further growth of his or her malignant B lymphocytes. After this therapy, patients are infused with their own or the donor's hematopoietic stem cells. One study reported a 3-year survival rate of 36% and another reported a median progression-free survival time (i.e. time disease does not worsen) of 11.2 months with a median overall survival time that was not reached after 54 months of follow-up.[26] A meta-analysis of 4 previous studies reported that 72 individuals receiving allogenic hematopoietic stem cell transplant (AHSCT) for RT (almost all cases were DLBCL-RT) achieved an overall response rate (percentage of patients showing a decrease in their disease) of 79%, complete response rate (percentage of patients showing an apparent disappearance of disease) of 33%, and 2-year and 5-year survival rates of 46 and 35%, respectively. Since the overall mean survival time in many studies had been less than 10 months for individuals not treated with AHSCT, the study concluded that AHSCT is a reasonable treatment option for fit individuals whose disease responded to ablation therapy.[27] However, these studies,[27] as well as other studies on homologous and/or autologous transplantation for DLBCL-RT, selected individuals for transplantation based on their partial or complete responses to ablation therapy[26] and often choose only patients who were physically and otherwise best fit to receive the transplant: in one study, only 20 of 148 individuals with DLBCL-RT were deemed eligible for transplantation.[1] Further studies, preferably randomized controlled trials, are needed to determine if this treatment regimen improves the outcome of such highly selected DLBCL-RT patients.
HL-RT treatment and prognosis
Historically, HL-RT cases were typically treated with regimens directed against CLL/SLL or other malignancies but not those used to treat Hodgkin's lymphomas not due to RT, i.e. HL-not RT. Median overall survival times in individual treated with these regimens varied between 0.8–3.9 years.[28] Currently, HL-RT is treated with chemotherapy regimens such as ABVD (i.e. adriamycin, bleomycin, vinblastine, and dacarbazine) which are used to treat HL-not RT. In a multicenter study, 62 individuals with HL-RT were treated with ABVD or an AVD-based regimen. These individuals had a median overall survival of 13.2 years, a time similar to that seen in most subtypes of HL-not RT when matched for patient age at the time of treatment. Hematopoietic stem cell transplantation in 7 of these individuals did not improve their median overall survival times.[28] Based on this result, one study concluded that stem cell transplantation given as consolidation therapy (i.e. therapy given to improve the gains from the preceding therapy) is not recommended to treat HL-RT.[1] In a study of 8 individuals with HL-RT treated with BEACOPP (2 cases), ABVD (1 case), or other regimens (5 cases) directed against HL-not RT, the median overall survival time was 7 years.[29] A retrospective review study of type 1 HL-RT cases (which have a poorer prognoses than type 2 HL-RT) found that individuals who received therapy regimens directed against HL-not RT had a median overall survival time of 57 months, significantly higher than those treated with regimens used to treat CLL/CSS (medium overall survival time of 8.4 months).[21] Currently, regimens, particularly ABVD, used to treat HL-not RT are the standard of care for treating HL-RT.[1][28]
References
- Sigmund AM, Kittai AS (August 2022). "Richter's Transformation". Current Oncology Reports. 24 (8): 1081–1090. doi:10.1007/s11912-022-01274-4. PMID 35384590.
- Musanhu E, Sharma RK, Attygalle A, Wotherspoon A, Chau I, Cunningham D, Dearden C, El-Sharkawi D, Iyengar S, Sharma B (November 2021). "Chronic lymphocytic leukaemia and Richter's transformation: multimodal review and new imaging paradigms". Clinical Radiology. 76 (11): 789–800. doi:10.1016/j.crad.2021.06.001. PMID 34217434.
- Rossi D, Gaidano G (March 2009). "Richter syndrome: molecular insights and clinical perspectives". Hematol Oncol. 27 (1): 1–10. doi:10.1002/hon.880. PMID 19206112.
- D'Addona M, Giudice V, Pezzullo L, Ciancia G, Baldi C, Gorrese M, Bertolini A, Campana A, Fresolone L, Manzo P, Zeppa P, Serio B, Selleri C (August 2022). "Hodgkin Lymphoma and Hairy Cell Leukemia Arising from Chronic Lymphocytic Leukemia: Case Reports and Literature Review". Journal of Clinical Medicine. 11 (16). doi:10.3390/jcm11164674. PMC 9410146. PMID 36012912.
- Trimech M, Letourneau A, Missiaglia E, De Prijck B, Nagy-Hulliger M, Somja J, Vivario M, Gaulard P, Lambert F, Bisig B, de Leval L (June 2021). "Angioimmunoblastic T-Cell Lymphoma and Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma: A Novel Form of Composite Lymphoma Potentially Mimicking Richter Syndrome". The American Journal of Surgical Pathology. 45 (6): 773–786. doi:10.1097/PAS.0000000000001646. PMID 33739791.
- Liu H, Miao Y, Ferrajoli A, Tang G, McDonnell T, Medeiros LJ, Hu S (March 2020). "Leukemic phase of Richter transformation: A mimic of acute myeloid leukemia that responded to Ibrutinib monotherapy". American Journal of Hematology. doi:10.1002/ajh.25782. PMID 32162729.
- Travis LB, Curtis RE, Hankey BF, Fraumeni JF (September 1992). "Second cancers in patients with chronic lymphocytic leukemia". Journal of the National Cancer Institute. 84 (18): 1422–7. doi:10.1093/jnci/84.18.1422. PMID 1512794.
- Kumar V, Ailawadhi S, Bojanini L, Mehta A, Biswas S, Sher T, Roy V, Vishnu P, Marin-Acevedo J, Alegria VR, Paulus A, Aulakh S, Iqbal M, Manochakian R, Tan W, Chanan-Khan A, Ailawadhi M (September 2019). "Trends in the risk of second primary malignancies among survivors of chronic lymphocytic leukemia". Blood Cancer Journal. 9 (10): 75. doi:10.1038/s41408-019-0237-1. PMC 6768881. PMID 31570695.
- Wąsik-Szczepanek E, Szymczyk A, Szczepanek D, Wszoła-Kleinrok J, Chocholska S, Pluta A, Hus M (December 2018). "Richter syndrome: A rare complication of chronic lymphocytic leukemia or small lymphocytic lymphoma". Advances in Clinical and Experimental Medicine : Official Organ Wroclaw Medical University. 27 (12): 1683–1689. doi:10.17219/acem/75903. PMID 30156387.
- Soilleux EJ, Wotherspoon A, Eyre TA, Clifford R, Cabes M, Schuh AH (December 2016). "Diagnostic dilemmas of high-grade transformation (Richter's syndrome) of chronic lymphocytic leukaemia: results of the phase II National Cancer Research Institute CHOP-OR clinical trial specialist haemato-pathology central review". Histopathology. 69 (6): 1066–1076. doi:10.1111/his.13024. PMID 27345622.
- Chiorazzi N, Chen SS, Rai KR (February 2021). "Chronic Lymphocytic Leukemia". Cold Spring Harbor Perspectives in Medicine. 11 (2). doi:10.1101/cshperspect.a035220. PMID 32229611.
- Tadmor T, Levy I (October 2021). "Richter Transformation in Chronic Lymphocytic Leukemia: Update in the Era of Novel Agents". Cancers. 13 (20). doi:10.3390/cancers13205141. PMC 8533993. PMID 34680290.
- Richter MN (July 1928). "Generalized Reticular Cell Sarcoma of Lymph Nodes Associated with Lymphatic Leukemia". The American Journal of Pathology. 4 (4): 285–292.7. PMC 2006994. PMID 19969796.
- LORTHOLARY P, BOIRON M, RIPAULT P, LEVY JP, MANUS A, BERNARD J (1964). "[CHRONIC LYMPHOID LEUKEMIA SECONDARILY ASSOCIATED WITH A MALIGNANT RETICULOPATHY: RICHTER'S SYNDROME]". Nouvelle Revue Francaise D'hematologie (in French). 4: 621–44. PMID 14199493.
- Lenartova A, Randen U, Johannesen TB, Tjønnfjord GE (June 2019). "Richter syndrome epidemiology in a large population based chronic lymphocytic leukemia cohort from Norway". Cancer Epidemiology. 60: 128–133. doi:10.1016/j.canep.2019.04.002. PMID 30986631.
- Parikh SA, Kay NE, Shanafelt TD (March 2014). "How we treat Richter syndrome". Blood. 123 (11): 1647–57. doi:10.1182/blood-2013-11-516229. PMC 3954047. PMID 24421328.
- Elnair R, Ellithi M, Kallam A, Shostrom V, Bociek RG (October 2021). "Outcomes of Richter's transformation of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL): an analysis of the SEER database". Annals of Hematology. 100 (10): 2513–2519. doi:10.1007/s00277-021-04603-y. PMID 34279675.
- Rhodes JM, Mato AR (July 2019). "PET/Computed Tomography in Chronic Lymphocytic Leukemia and Richter Transformation". PET Clinics. 14 (3): 405–410. doi:10.1016/j.cpet.2019.03.007. PMID 31084779.
- Ravindran A, Kurtin PJ, King RL, Yuan J, Feldman AL, Rech KL, McPhail ED, Parikh SA, Ding W, Shi M (July 2022). "Aberrant expression of lymphoid enhancer-binding factor 1 in Hodgkin lymphoma". Human Pathology. 125: 2–10. doi:10.1016/j.humpath.2022.04.004. PMID 35421421.
- Xiao W, Chen WW, Sorbara L, Davies-Hill T, Pittaluga S, Raffeld M, Jaffe ES (September 2016). "Hodgkin lymphoma variant of Richter transformation: morphology, Epstein-Barr virus status, clonality, and survival analysis-with comparison to Hodgkin-like lesion". Human Pathology. 55: 108–116. doi:10.1016/j.humpath.2016.04.019. PMC 4981556. PMID 27184478.
- King RL, Gupta A, Kurtin PJ, Ding W, Call TG, Rabe KG, Kenderian SS, Leis JF, Wang Y, Schwager SM, Slager SL, Kay NE, Koehler A, Ansell SM, Inwards DJ, Habermann TM, Shi M, Hanson CA, Howard MT, Parikh SA (January 2022). "Chronic lymphocytic leukemia (CLL) with Reed-Sternberg-like cells vs Classic Hodgkin lymphoma transformation of CLL: does this distinction matter?". Blood Cancer Journal. 12 (1): 18. doi:10.1038/s41408-022-00616-6. PMC 8799721. PMID 35091549.
- Rezk SA, Zhao X, Weiss LM (June 2018). "Epstein–Barr virus–associated lymphoid proliferations, a 2018 update". Human Pathology. 79: 18–41. doi:10.1016/j.humpath.2018.05.020. PMID 29885408. S2CID 47010934.
- Gamaleldin MA, Ghallab OM, Nadwan EA, Abo Elwafa RA (November 2021). "PD-1 and PD-L1 gene expressions and their association with Epstein-Barr virus infection in chronic lymphocytic leukemia". Clinical & Translational Oncology : Official Publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico. 23 (11): 2309–2322. doi:10.1007/s12094-021-02657-y. PMID 34120295.
- Gángó A, Kiss R, Farkas P, Hanna E, Demeter J, Deák B, Lévai D, Kotmayer L, Alpár D, Matolcsy A, Bödör C, Mátrai Z, Timár B (February 2022). "Morphologic and molecular analysis of Richter syndrome in chronic lymphocytic leukaemia patients treated with ibrutinib or venetoclax". Pathology. 54 (1): 95–103. doi:10.1016/j.pathol.2021.04.008. PMID 34332791.
- Reed A, Sommerhalder D (December 2019). "The Use of R-Hyper-CVAD in a Rare Case of Primary Bone Marrow Diffuse Large B-Cell Lymphoma". Journal of Hematology. 8 (4): 165–167. doi:10.14740/jh559. PMC 7155813. PMID 32300465.
- Kim HT, Baker PO, Parry E, Davids M, Alyea EP, Ho VT, Cutler C, Koreth J, Gooptu M, Romee R, Nikiforow S, Antin JH, Ritz J, Soiffer RJ, Wu CJ, Brown JR (December 2021). "Allogeneic hematopoietic cell transplantation outcomes in patients with Richter's transformation". Haematologica. 106 (12): 3219–3222. doi:10.3324/haematol.2021.279033. PMC 8634179. PMID 34435483.
- Aulakh S, Reljic T, Yassine F, Ayala E, Chavez JC, Chanan-Khan A, Pinilla-Ibarz J, Kumar A, Kharfan-Dabaja MA (March 2021). "Allogeneic hematopoietic cell transplantation is an effective treatment for patients with Richter syndrome: A systematic review and meta-analysis". Hematology/oncology and Stem Cell Therapy. 14 (1): 33–40. doi:10.1016/j.hemonc.2020.05.002. PMC 7666647. PMID 32473105.
- Stephens DM, Boucher K, Kander E, Parikh SA, Parry EM, Shadman M, Pagel JM, Cooperrider J, Rhodes J, Mato A, Winter A, Hill B, Gaballa S, Danilov A, Phillips T, Brander DM, Smith SM, Davids M, Rogers K, Glenn MJ, Byrd JC (November 2021). "Hodgkin lymphoma arising in patients with chronic lymphocytic leukemia: outcomes from a large multi-center collaboration". Haematologica. 106 (11): 2845–2852. doi:10.3324/haematol.2020.256388. PMC 8561295. PMID 33054118.
- Al-Sawaf O, Robrecht S, Bahlo J, Fink AM, Cramer P, V Tresckow J, Lange E, Kiehl M, Dreyling M, Ritgen M, Dürig J, Tausch E, Schneider C, Stilgenbauer S, Wendtner CM, Fischer K, Goede, Hallek M, Eichhorst B (January 2021). "Richter transformation in chronic lymphocytic leukemia (CLL)-a pooled analysis of German CLL Study Group (GCLLSG) front line treatment trials". Leukemia. 35 (1): 169–176. doi:10.1038/s41375-020-0797-x. PMID 32203141.