Vitelliform macular dystrophy
Vitelliform macular dystrophy is an irregular autosomal dominant eye disorder which can cause progressive vision loss.[1] This disorder affects the retina, specifically cells in a small area near the center of the retina called the macula. The macula is responsible for sharp central vision, which is needed for detailed tasks such as reading, driving, and recognizing faces. The condition is characterized by yellow (or orange), slightly elevated, round structures similar to the yolk (Latin vitellus) of an egg.[2]
| Vitelliform macular dystrophy | |
|---|---|
| Other names | Vitelliform dystrophy |
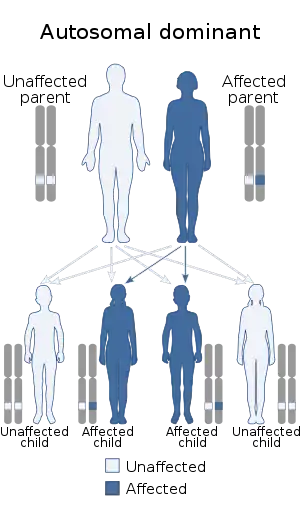 | |
| Best disease, the early-onset form of vitelliform macular dystrophy, has an autosomal dominant pattern of inheritance. | |
| Specialty | Ophthalmology, medical genetics |
Genetics
Best disease is inherited in an autosomal dominant pattern,[3] which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition.
The inheritance pattern of adult-onset vitelliform macular dystrophy is definitively autosomal dominant.[2] Many affected people, however, have no history of the disorder in their family and only a small number of affected families have been reported. This is because the penetrance of the condition is incomplete; therefore, it is possible for an individual to have a copy of the mutant allele and not display the VMD phenotype. The ratio of males to females is approximately 1:1.[2]
Pathophysiology
Mutations in the RDS and VMD2 genes cause vitelliform macular dystrophy. Mutations in the VMD2 gene are responsible for Best disease. Changes in either the VMD2 or RDS gene can cause the adult-onset form of vitelliform macular dystrophy; however, fewer than a quarter of cases result from mutations in these two genes. In most cases, the cause of the adult-onset form is unknown.
The VMD2 gene provides instructions for making a protein called bestrophin.[4] Although its exact function is uncertain, this protein likely acts as a channel that controls the movement of negatively charged chlorine atoms (chloride ions) into or out of cells in the retina. Mutations in the VMD2 gene probably lead to the production of an abnormally shaped channel that cannot regulate the flow of chloride. Researchers have not determined how these malfunctioning channels are related to the buildup of lipofuscin in the macula and progressive vision loss.
The RDS gene provides instructions for making a protein called peripherin.[5] This protein is essential for the normal function of light-sensing (photoreceptor) cells in the retina. Mutations in the RDS gene disrupt the structures in these cells that contain light-sensing pigments, leading to vision loss. It is unclear why RDS mutations affect only central vision in people with adult-onset vitelliform macular dystrophy.
Diagnosis
Vitelliform macular dystrophy causes a fatty yellow pigment (lipofuscin) to build up in cells underlying the macula.[6] The retinal pigment epithelium also degenerates. Over time, the abnormal accumulation of this substance can damage the cells that are critical for clear central vision. As a result, people with this disorder often lose their central vision and may experience blurry or distorted vision, and loss is rarely symmetric. Scotomata appear, first with red light and then for green; finally, relative (or in more serious cases, absolute) scotomata occur with white light. Vitelliform macular dystrophy does not affect side (peripheral) vision or the ability to see at night.
Researchers have described two forms of vitelliform macular dystrophy with similar features. The early-onset form (known as Best disease) usually appears in childhood.[7] The disease received its name from a German ophthalmologist Friedrich Best who defined a pedigree living with various stages of the disease in 1905. The onset of symptoms and the severity of vision loss vary widely. The adult-onset form begins later, usually in middle age, and tends to cause relatively mild vision loss. The two forms of vitelliform macular dystrophy each have characteristic changes in the macula that can be detected during an eye examination.
In the Best disease, the paraclinic evaluations such as fundus fluorescein angiography or indocyanine green angiography, along with baseline and final electrooculography (EOG) results, may shed the light on the progression of disease.[8][9]
References
- Musarella MA (May 2001). "Molecular genetics of macular degeneration". Documenta Ophthalmologica. Advances in Ophthalmology. 102 (3): 165–77. doi:10.1023/A:1017510515893. PMID 11556484.
- Deutman A, Hoyng C, van Lith-Verhoeven J (2006). "Macular dystrophies". Retina (4 ed.). Elsevier Mosby. pp. 1177–81.
- Heidary F, Gharebaghi R (March 2021). "Natural course of the vitelliform stage in best vitelliform macular dystrophy: a five-year follow-up study". Graefe's Archive for Clinical and Experimental Ophthalmology = Albrecht von Graefes Archiv für Klinische und Experimentelle Ophthalmologie. 259 (3): 787–788. doi:10.1007/s00417-020-04888-1. PMID 32785779.
- Marmorstein AD, Marmorstein LY, Rayborn M, Wang X, Hollyfield JG, Petrukhin K (November 2000). "Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium". Proceedings of the National Academy of Sciences of the United States of America. 97 (23): 12758–63. doi:10.1073/pnas.220402097. PMC 18837. PMID 11050159.
- Kohl S, Giddings I, Besch D, Apfelstedt-Sylla E, Zrenner E, Wissinger B (1998). "The role of the peripherin/RDS gene in retinal dystrophies". Acta Anatomica. 162 (2–3): 75–84. doi:10.1159/000046471. PMID 9831753.
- Guziewicz KE, Sinha D, Gómez NM, Zorych K, Dutrow EV, Dhingra A, et al. (May 2017). "Bestrophinopathy: An RPE-photoreceptor interface disease". Progress in Retinal and Eye Research. 58: 70–88. doi:10.1016/j.preteyeres.2017.01.005. PMC 5441932. PMID 28111324.
- Heidary F, Hitam WH, Ngah NF, George TM, Hashim H, Shatriah I (March 2011). "Intravitreal Ranibizumab for Choroidal Neovascularization in Best's Vitelliform Macular Dystrophy in a 6-Year-Old Boy". Journal of Pediatric Ophthalmology and Strabismus. 48 Online (6): e19-22. doi:10.3928/01913913-20110308-02. PMID 21417187.
- Heidary F, Gharebaghi R (March 2021). "Natural course of the vitelliform stage in best vitelliform macular dystrophy: a five-year follow-up study". Graefe's Archive for Clinical and Experimental Ophthalmology = Albrecht von Graefes Archiv für Klinische und Experimentelle Ophthalmologie. 259 (3): 787–788. doi:10.1007/s00417-020-04888-1. PMID 32785779.
- Orellana J, Friedman AH (1993). Best’s Disease. Clinico-Pathological Atlas of Congenital Fundus Disorders. New York, NY: Springer. pp. 147–150. doi:10.1007/978-1-4613-9320-7_32. ISBN 978-1-4613-9322-1.
Further reading
- MacDonald IM, Lee T (December 2003). "Best Vitelliform Macular Dystrophy". In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A, MacDonald IM, Lee T (eds.). GeneReviews. Seattle (WA): University of Washington, Seattle. PMID 20301346.
- George Priya Doss C, Chakraborty C, Monford Paul Abishek N, Thirumal Kumar D, Narayanan V (2014). "Application of evolutionary based in silico methods to predict the impact of single amino acid substitutions in vitelliform macular dystrophy". Advances in Protein Chemistry and Structural Biology. 94: 177–267. doi:10.1016/B978-0-12-800168-4.00006-8. PMID 24629188.