Channelopathy
Channelopathies are a group of diseases caused by the dysfunction of ion channel subunits or their interacting proteins. These diseases can be inherited or acquired by other disorders, drugs, or toxins. Mutations in genes encoding ion channels, which impair channel function, are the most common cause of channelopathies.[1] There are more than 400 genes that encode ion channels, found in all human cell types and are involved in almost all physiological processes.[2] Each type of channel is a multimeric complex of subunits encoded by a number of genes. Depending where the mutation occurs it may affect the gating, conductance, ion selectivity, or signal transduction of the channel.
| Channelopathy | |
|---|---|
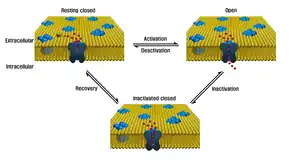 | |
| Sodium channel, implicated in channelopathies including Brugada syndrome, Long QT syndrome, Dravet syndrome, Paramyotonia congenita | |
| Specialty | Medical genetics, Neuromuscular medicine, Cardiology |
| Symptoms | Dependent on type. Include: Syncope, muscle weakness, seizures, breathlessness |
| Complications | Dependent on type. Include: Sudden death |
| Causes | Genetic variants |
Channelopathies can be categorized based on the organ system which they are associated with. In the cardiovascular system, the electrical impulse needed for each heartbeat is made possible by the electrochemical gradient of each heart cell. Because the heartbeat is dependent on the proper movement of ions across the surface membrane, cardiac channelopathies make up a key group of heart diseases.[3] Long QT syndrome, the most common form of cardiac channelopathy, is characterized by prolonged ventricular repolarization, predisposing to a high risk of ventricular tachyarrhythmias (e.g., torsade de pointes), syncope, and sudden cardiac death.[1]
The channelopathies of human skeletal muscle include hyper- and hypokalemic (high and low potassium blood concentrations) periodic paralysis, myotonia congenita and paramyotonia congenita.
Channelopathies affecting synaptic function are a type of synaptopathy.
Causes
Genetic type
Mutations in genes encoding ion channels, which cause defects in channel function, are the most common cause of channelopathies.[1]
Acquired type
Acquired channelopathies are caused by acquired disorders, drug use, toxins, etc.[1]
Types
The types in the following table are commonly accepted. Channelopathies currently under research, like Kir4.1 potassium channel in multiple sclerosis, are not included.
References
- Kim JB (January 2014). "Channelopathies". Korean Journal of Pediatrics. 57 (1): 1–18. doi:10.3345/kjp.2014.57.1.1. PMC 3935107. PMID 24578711.
- Imbrici P, Liantonio A, Camerino GM, De Bellis M, Camerino C, Mele A, et al. (2016-05-10). "Therapeutic Approaches to Genetic Ion Channelopathies and Perspectives in Drug Discovery". Frontiers in Pharmacology. 7: 121. doi:10.3389/fphar.2016.00121. PMC 4861771. PMID 27242528.
- Marbán E (January 2002). "Cardiac channelopathies". Nature. 415 (6868): 213–218. Bibcode:2002Natur.415..213M. doi:10.1038/415213a. PMID 11805845. S2CID 4419017.
- Vargas-Alarcon G, Alvarez-Leon E, Fragoso JM, Vargas A, Martinez A, Vallejo M, Martinez-Lavin M (February 2012). "A SCN9A gene-encoded dorsal root ganglia sodium channel polymorphism associated with severe fibromyalgia". BMC Musculoskeletal Disorders. 13: 23. doi:10.1186/1471-2474-13-23. PMC 3310736. PMID 22348792.
- Smith RS, Kenny CJ, Ganesh V, Jang A, Borges-Monroy R, Partlow JN, et al. (September 2018). "Sodium Channel SCN3A (NaV1.3) Regulation of Human Cerebral Cortical Folding and Oral Motor Development". Neuron. 99 (5): 905–913.e7. doi:10.1016/j.neuron.2018.07.052. PMC 6226006. PMID 30146301.
- Smith RS, Florio M, Akula SK, Neil JE, Wang Y, Hill RS, et al. (June 2021). "Early role for a Na+,K+-ATPase (ATP1A3) in brain development". Proceedings of the National Academy of Sciences of the United States of America. 118 (25): e2023333118. doi:10.1073/pnas.2023333118. PMC 8237684. PMID 34161264.
- Simons C, Rash LD, Crawford J, Ma L, Cristofori-Armstrong B, Miller D, et al. (January 2015). "Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy". Nature Genetics. 47 (1): 73–77. doi:10.1038/ng.3153. PMID 25420144. S2CID 52799681.
- Hunter JV, Moss AJ (January 2009). "Seizures and arrhythmias: Differing phenotypes of a common channelopathy?". Neurology. 72 (3): 208–209. doi:10.1212/01.wnl.0000339490.98283.c5. PMID 19153369. S2CID 207103822.
- Mulley JC, Scheffer IE, Petrou S, Berkovic SF (April 2003). "Channelopathies as a genetic cause of epilepsy". Current Opinion in Neurology. 16 (2): 171–176. doi:10.1097/00019052-200304000-00009. PMID 12644745. S2CID 40441842.
Bibliography
- Song YW, Kim SJ, Heo TH, Kim MH, Kim JB (December 2012). "Normokalemic periodic paralysis is not a distinct disease". Muscle & Nerve. 46 (6): 914–916. doi:10.1002/mus.23441. PMID 22926674. S2CID 43821573.
External links
VIDEO Channel Surfing in Pediatrics by Carl E. Stafstrom, M.D., at the UW-Madison Health Sciences Learning Center.
- "The Weiss Lab". The Weiss Lab is investigating the molecular and cellular mechanisms underlying human diseases caused by dysfunction of ion channels.
- The Channelopathy Foundation - Foundation for Ion Channel diseases
- Cystic Fibrosis Foundation
- Rare Diseases Clinical Research Network