Young–Madders syndrome
Young–Madders syndrome, alternatively known as Pseudotrisomy 13 syndrome or holoprosencephaly–polydactyly syndrome, is a genetic disorder resulting from defective and duplicated chromosomes which result in holoprosencephaly, polydactyly, facial malformations and intellectual disability, with a significant variance in the severity of symptoms being seen across known cases.[1] Many cases often suffer with several other genetic disorders, and some have presented with hypoplasia, cleft lip, cardiac lesions and other heart defects. In one case in 1991 and another in 2000 the condition was found in siblings who were the product of incest. Many cases are diagnosed prenatally and often in siblings.[1] Cases are almost fatal in the prenatal stage with babies being stillborn.
| Young–Madders syndrome | |
|---|---|
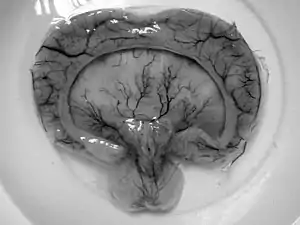 | |
| Gross pathology specimen from a case of alobar holoprosencephaly, a clinical manifestation of Young–Madders syndrome first described as a new condition by doctors Young and Madders in 1987. |
Though it is now thought that earlier cases were misdiagnosed as other genetic disorders with similar pathology—such as Smith–Lemli–Opitz syndrome—the earliest publicised recognition of the condition as a new, hitherto unclassified, genetic disorder was made by two British doctors in Leicester in 1987. Though they identified the condition, later named for them, they did not identify the genetic anomalies responsible but suspected a link with trisomy 13 due to the similar symptoms. With only one or two occurrences documented towards the end of the decade, a group of eight doctors published a five-patient case-study in 1991 which identified the likely chromosomal factors that caused the condition, similar to but distinct from trisomy 13, and gave it the name 'holoprosencephaly–polydactyly syndrome' based on its two most prolific presenting conditions. Later research showed that the condition could manifest in patients with normal karyotypes, without duplication of the chromosomes, and the most recent genetic research implicates problems with the gene code FBXW11 as a likely cause.
Presentation
Young–Madders syndrome is detectable from the fetal stage of development largely due to the distinctive consequences of holoprosencephaly, a spectrum of defects or malformations of the brain and face. Facial defects which may manifest in the eyes, nose, and upper lip, featuring cyclopia, anosmia, or in the growth of only a single central incisor, and severe overlapping of the bones of the skull.[2][3] Cardiac and in some cases pulmonary deformities are present.[2] Another signature deformity is bilateral polydactyly, and many patients also suffer from hypoplasia and genital deformities.[1]
Diagnosis
History
Early cases
A publication in the Journal of Medical Genetics in 1987 by Dr. I. Young and D. Madders of Leicester Royal Infirmary in the United Kingdom described the then-unknown condition when presenting "a stillborn male infant with pre-maxillary agenesis, bilateral microphthalmos, alobar holoprosencephaly, hydrocephalus, ventricular and atrial septal defects, small penis, bilateral cryptorchidism, and bilateral upper limb postaxial polydactyly."[2] Both doctors noted no use of drugs, alcohol or cigarettes by the mother, and the baby was delivered normally after forty-one weeks of gestation. It was the first child of the parents, who were not related and went on to have another child successfully however this child was a stillbirth. There was severe overlapping of the bones of the skull and a cleft lip in addition to the bilateral polydactyly. Of the organs, Young and Madders noted missing parts of the tricuspid valve and other small cardiac defects, as well as the holoprosencephaly.[2] Both doctors consulted various medical databases and, after discounting Meckel syndrome due to a lack of renal abnormalities, concluded that this was a hitherto unclassified condition. After later classification, it was later named for the two doctors,[2] though at the time of publication it was termed 'pseudotrisomy 13' due to similarities with the condition Trisomy 13.[1] Another case in 1989 with similar symptoms was also published as an example of 'pseudotrisomy 13', and there was no evidence of an extra chromosome, further suggesting that Trisomy 13 was a separate condition.[1]
Identifying chromosome thirteen

Sporadic reports of the case continued, with 'pseudotrisomy 13' becoming a common term due to the similar pathology to Trisomy 13. However, there was a growing belief that unlike Trisomy 13, Young–Madders syndrome was not caused by a duplicated chromosome, and in fact the cause lay in some other fault with chromosome thirteen. In 1991 a publication in the Journal of Medical Genetics by a group of eight doctors, based on a five-patient case-study, argued that Trisomy 13 and Young–Madders syndrome were two distinct conditions and renamed the disorder to avoid confusion. Their case studies, when viewed together, suggested a recessive genetic cause,[4]
based on the repeated instances of holoprosencephaly polydactyly in the aforementioned five cases, which led to the suspicion of an anomaly in chromosome 13's genetic coding.[4] Chromosome thirteen spans about 114 million base pairs (the building material of DNA) and represents between 3.5 and 4% of the total DNA in cells. Problems with this chromosome account for several conditions including nonsyndromic deafness, Waardenburg syndrome and Wilson's disease.[4]
The majority of the cases discussed in the journal were still born, with death occurring between twenty-six and thirty-four weeks of gestation. All suffered with the features of Young–Madders syndrome, with varying cardiac problems and facial deformities. The distinctive bilateral polydactyly and overlapping of the cranial skull plates were present, though some had no deformities in their internal organs while others had lung deformities alone. Hydrocephalus and holoprosencephaly were present in all.[4] The publication noted the work of Young and Madders and suggested that the cases were linked, and also identified two cases from a year previously - 1986 - which had until then been diagnosed as Smith–Lemli–Opitz syndrome. The doctors discounted several other similar genetic conditions including Varadi–Papp syndrome and Grote syndrome, and discarded the term 'pseudotrisomy 13 syndrome' as misleading, preferring 'holoprosencephaly-polydactyly syndrome'.[4]
Further research
After the publications by Verloes, A., S. Aymé et al. 'holoprosencephaly-polydactyly syndrome' became the preferred term. Doctors Hennekam and Van Noort published later in 1991 an account of further case studies involving a brother and sister which emphasised the involvement of genetic heart defects. They also noted that the parents were second-cousins.[5] A pair of siblings with holoprosencephaly and hypoplasia were recognised in 1993, with one of the siblings having polydactyly.[6] By 1994 a sufferer had been identified with a median cleft lip, cardiac and genital deformities which matched those found by Young and Madders. A review of twenty-two reported cases took place, finding 20 instances of polydactyly. Blood tests and karyotype were normal, further evidencing a distinction between the condition and that of Trisomy 13.[7] In 2000 another case was published were the parents were first cousins, thus providing more evidence that the condition was autosomal recessive.[8] In 2006 a report in the Journal of Human Genetics on a male patient with all the symptoms of Young–Madders syndrome - including severe facial deformities and mild intellectual disability - built upon a 1993 genetic study to identify duplicate codings and other anomalies on gene FBXW11 as a possible cause of the condition.[9]
References
- "Pseudotrisomy 13 Syndrome". OMIM. 264480. Retrieved 25 January 2014.
- Young ID, Madders DJ (November 1987). "Unknown syndrome: holoprosencephaly, congenital heart defects, and polydactyly". Journal of Medical Genetics. 24 (11): 714–715. doi:10.1136/jmg.24.11.714. PMC 1050355. PMID 3430550.
- Totori-Donati P, Rossi A, Biancheri R (2005). "Brain Malformations". In Totori-Donati P, Rossi A, Raybaud C (eds.). Pediatric Neuroradiology: Brain, Head, Neck and Spine. Vol. 1. Springer. pp. 92–95. ISBN 3-540-41077-5.
- Verloes A, Aymé S, Gambarelli D, Gonzales M, Le Merrer M, Mulliez N, et al. (May 1991). "Holoprosencephaly-polydactyly ('pseudotrisomy 13') syndrome: a syndrome with features of hydrolethalus and Smith-Lemli-Opitz syndromes. A collaborative multicentre study". Journal of Medical Genetics. 28 (5): 297–303. doi:10.1136/jmg.28.5.297. PMC 1016846. PMID 1865466.
- Hennekam RC, van Noort G, de la Fuente AA (November 1991). "Familial holoprosencephaly, heart defects, and polydactyly". American Journal of Medical Genetics. 41 (2): 258–262. doi:10.1002/ajmg.1320410226. PMID 1785646.
- Seller MJ, Chitty LS, Dunbar H (November 1993). "Pseudotrisomy 13 and autosomal recessive holoprosencephaly". Journal of Medical Genetics. 30 (11): 970–971. doi:10.1136/jmg.30.11.970. PMC 1016613. PMID 8301659.
- Ramos-Arroyo MA, de Miguel C, Valiente A, Moreno-Laguna S (April 1994). "Further delineation of pseudotrisomy 13 syndrome: a case without polydactyly". American Journal of Medical Genetics. 50 (2): 177–179. doi:10.1002/ajmg.1320500208. PMID 8010349.
- Amor DJ, Woods CG (April 2000). "Pseudotrisomy 13 syndrome in siblings". Clinical Dysmorphology. 9 (2): 115–118. doi:10.1097/00019605-200009020-00008. PMID 10826623.
- Koolen DA, Herbergs J, Veltman JA, Pfundt R, van Bokhoven H, Stroink H, et al. (2006). "Holoprosencephaly and preaxial polydactyly associated with a 1.24 Mb duplication encompassing FBXW11 at 5q35.1". Journal of Human Genetics. 51 (8): 721–726. doi:10.1007/s10038-006-0010-8. PMID 16865294.