Johnson–Corey–Chaykovsky reaction
| Johnson-Corey–Chaykovsky reaction | |
|---|---|
| Named after | A. William Johnson Elias James Corey Michael Chaykovsky |
| Reaction type | Ring forming reaction |
| Identifiers | |
| Organic Chemistry Portal | corey-chaykovsky-reaction |
The Johnson–Corey–Chaykovsky reaction (sometimes referred to as the Corey–Chaykovsky reaction or CCR) is a chemical reaction used in organic chemistry for the synthesis of epoxides, aziridines, and cyclopropanes. It was discovered in 1961 by A. William Johnson and developed significantly by E. J. Corey and Michael Chaykovsky. The reaction involves addition of a sulfur ylide to a ketone, aldehyde, imine, or enone to produce the corresponding 3-membered ring. The reaction is diastereoselective favoring trans substitution in the product regardless of the initial stereochemistry. The synthesis of epoxides via this method serves as an important retrosynthetic alternative to the traditional epoxidation reactions of olefins.
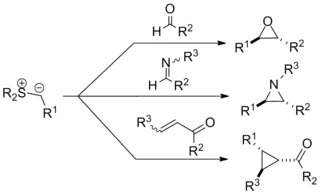
The reaction is most often employed for epoxidation via methylene transfer, and to this end has been used in several notable total syntheses (See Synthesis of epoxides below). Additionally detailed below are the history, mechanism, scope, and enantioselective variants of the reaction. Several reviews have been published.[1][2][3][4][5][6]
History
The original publication by Johnson concerned the reaction of 9-dimethylsulfonium fluorenylide with substituted benzaldehyde derivatives. The attempted Wittig-like reaction failed and a benzalfluorene oxide was obtained instead, noting that "Reaction between the sulfur ylid and benzaldehydes did not afford benzalfluorenes as had the phosphorus and arsenic ylids."[7]
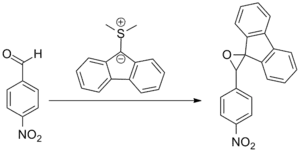
The subsequent development of (dimethyloxosulfaniumyl)methanide, (CH3)2SOCH2 and (dimethylsulfaniumyl)methanide, (CH3)2SCH2 (known as Corey–Chaykovsky reagents) by Corey and Chaykovsky as efficient methylene-transfer reagents established the reaction as a part of the organic canon.[8]
Mechanism
The reaction mechanism for the Johnson–Corey–Chaykovsky reaction consists of nucleophilic addition of the ylide to the carbonyl or imine group. A negative charge is transferred to the heteroatom and because the sulfonium cation is a good leaving group it gets expelled forming the ring. In the related Wittig reaction, the formation of the much stronger phosphorus-oxygen double bond prevents oxirane formation and instead, olefination takes place through a 4-membered cyclic intermediate.[4]

The trans diastereoselectivity observed results from the reversibility of the initial addition, allowing equilibration to the favored anti betaine over the syn betaine. Initial addition of the ylide results in a betaine with adjacent charges; density functional theory calculations have shown that the rate-limiting step is rotation of the central bond into the conformer necessary for backside attack on the sulfonium.[1]
_V2.png.webp)
The degree of reversibility in the initial step (and therefore the diastereoselectivity) depends on four factors, with greater reversibility corresponding to higher selectivity:[1]
- Stability of the substrate with higher stability leading to greater reversibility by favoring the starting material over the betaine.
- Stability of the ylide with higher stability similarly leading to greater reversibility.
- Steric hindrance in the betaine with greater hindrance leading to greater reversibility by disfavoring formation of the intermediate and slowing the rate-limiting rotation of the central bond.
- Solvation of charges in the betaine by counterions such as lithium with greater solvation allowing more facile rotation in the betaine intermediate, lowering the amount of reversibility.
Scope
The application of the Johnson–Corey–Chaykovsky reaction in organic synthesis is diverse. The reaction has come to encompass reactions of many types of sulfur ylides with electrophiles well beyond the original publications. It has seen use in a number of high-profile total syntheses, as detailed below, and is generally recognized as a powerful transformative tool in the organic repertoire.
Types of ylides
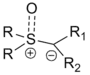
Many types of ylides can be prepared with various functional groups both on the anionic carbon center and on the sulfur. The substitution pattern can influence the ease of preparation for the reagents (typically from the sulfonium halide, e.g. trimethylsulfonium iodide) and overall reaction rate in various ways. The general format for the reagent is shown on the right.[1]
Use of a sulfoxonium allows more facile preparation of the reagent using weaker bases as compared to sulfonium ylides. (The difference being that a sulfoxonium contains a doubly bonded oxygen whereas the sulfonium does not.) The former react slower due to their increased stability. In addition, the dialkylsulfoxide by-products of sulfoxonium reagents are greatly preferred to the significantly more toxic, volatile, and odorous dialkylsulfide by-products from sulfonium reagents.[1]
The vast majority of reagents are monosubstituted at the ylide carbon (either R1 or R2 as hydrogen). Disubstituted reagents are much rarer but have been described:[1]
- If the ylide carbon is substituted with an electron-withdrawing group (EWG), the reagent is referred to as a stabilized ylide. These, similarly to sulfoxonium reagents, react much slower and are typically easier to prepare. These are limited in their usefulness as the reaction can become prohibitively sluggish: examples involving amides are widespread, with many fewer involving esters and virtually no examples involving other EWG's. For these, the related Darzens reaction is typically more appropriate.
- If the ylide carbon is substituted with an aryl or allyl group, the reagent is referred to as a semi-stabilized ylide. These have been developed extensively, second only to the classical methylene reagents (R1=R2=H). The substitution pattern on aryl reagents can heavily influence the selectivity of the reaction as per the criteria above.
- If the ylide carbon is substituted with an alkyl group the reagent is referred to as an unstabilized ylide. The size of the alkyl groups are the major factors in selectivity with these reagents.
The R-groups on the sulfur, though typically methyls, have been used to synthesize reagents that can perform enantioselective variants of the reaction (See Variations below). The size of the groups can also influence diastereoselectivity in alicyclic substrates.[1]
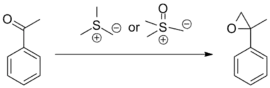
Synthesis of epoxides
Reactions of sulfur ylides with ketones and aldehydes to form epoxides are by far the most common application of the Johnson–Corey–Chaykovsky reaction. Examples involving complex substrates and 'exotic' ylides have been reported, as shown below.[9][10]
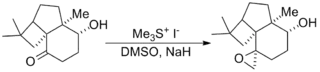
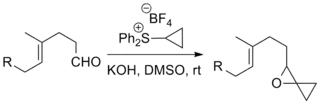
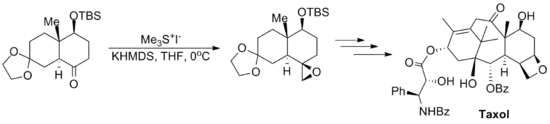
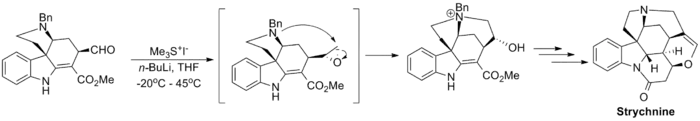
The reaction has been used in a number of notable total syntheses including the Danishefsky Taxol total synthesis, which produces the chemotherapeutic drug taxol, and the Kuehne Strychnine total synthesis which produces the pesticide strychnine.[11][12]
Synthesis of aziridines
The synthesis of aziridines from imines is another important application of the Johnson–Corey–Chaykovsky reaction and provides an alternative to amine transfer from oxaziridines. Though less widely applied, the reaction has a similar substrate scope and functional group tolerance to the carbonyl equivalent. The examples shown below are representative; in the latter, an aziridine forms in situ and is opened via nucleophilic attack to form the corresponding amine.[3][9]
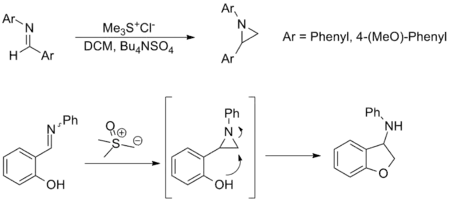
Synthesis of cyclopropanes
For addition of sulfur ylides to enones, higher 1,4-selectivity is typically obtained with sulfoxonium reagents than with sulfonium reagents. Many electron-withdrawing groups have been shown compatible with the reaction including ketones, esters, and amides (the example below involves a Weinreb amide). With further conjugated systems 1,6-addition tends to predominate over 1,4-addition.[3][9]
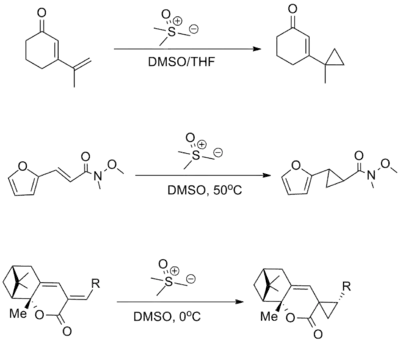
Other reactions
In addition to the reactions originally reported by Johnson, Corey, and Chaykovsky, sulfur ylides have been used for a number of related homologation reactions that tend to be grouped under the same name.
- With epoxides and aziridines the reaction serves as a ring-expansion to produce the corresponding oxetane or azetidine. The long reaction times required for these reactions prevent them from occurring as significant side reactions when synthesizing epoxides and aziridines.[9]
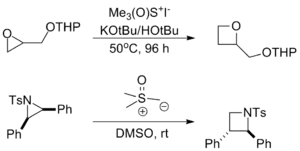
- Several cycloadditions wherein the ylide serves as a "nucleophilic carbenoid equivalent" have been reported.[9]
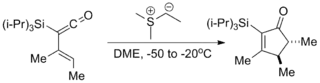
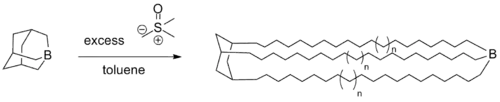
- Living polymerizations using trialkylboranes as the catalyst and (dimethyloxosulfaniumyl)methanide as the monomer have been reported for the synthesis of various complex polymers.[13]
Enantioselective variations
The development of an enantioselective (i.e. yielding an enantiomeric excess, which is labelled as "ee") variant of the Johnson–Corey–Chaykovsky reaction remains an active area of academic research. The use of chiral sulfides in a stoichiometric fashion has proved more successful than the corresponding catalytic variants, but the substrate scope is still limited in all cases. The catalytic variants have been developed almost exclusively for enantioselective purposes; typical organosulfide reagents are not prohibitively expensive and the racemic reactions can be carried out with equimolar amounts of ylide without raising costs significantly. Chiral sulfides, on the other hand, are more costly to prepare, spurring the advancement of catalytic enantioselective methods.[2]
Stoichiometric reagents
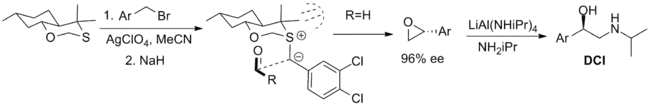
The most successful reagents employed in a stoichiometric fashion are shown below. The first is a bicyclic oxathiane that has been employed in the synthesis of the β-adrenergic compound dichloroisoproterenol (DCI) but is limited by the availability of only one enantiomer of the reagent. The synthesis of the axial diastereomer is rationalized via the 1,3-anomeric effect which reduces the nucleophilicity of the equatorial lone pair. The conformation of the ylide is limited by transannular strain and approach of the aldehyde is limited to one face of the ylide by steric interactions with the methyl substituents.[5][2]
The other major reagent is a camphor-derived reagent developed by Varinder Aggarwal of the University of Bristol. Both enantiomers are easily synthesized, although the yields are lower than for the oxathiane reagent. The ylide conformation is determined by interaction with the bridgehead hydrogens and approach of the aldehyde is blocked by the camphor moiety. The reaction employs a phosphazene base to promote formation of the ylide.[5][2]
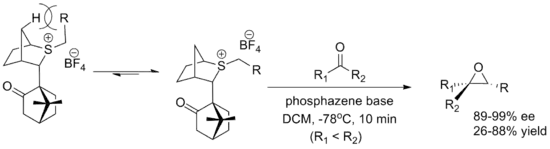
Catalytic reagents
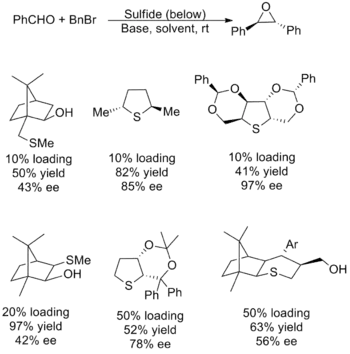
Catalytic reagents have been less successful, with most variations suffering from poor yield, poor enantioselectivity, or both. There are also issues with substrate scope, most having limitations with methylene transfer and aliphatic aldehydes. The trouble stems from the need for a nucleophilic sulfide that efficiently generates the ylide which can also act as a good leaving group to form the epoxide. Since the factors underlying these desiderata are at odds, tuning of the catalyst properties has proven difficult. Shown below are several of the most successful catalysts along with the yields and enantiomeric excess for their use in synthesis of (E)-stilbene oxide.[5][2]
Aggarwal has developed an alternative method employing the same sulfide as above and a novel alkylation involving a rhodium carbenoid formed in situ. The method too has limited substrate scope, failing for any electrophiles possessing basic substituents due to competitive consumption of the carbenoid.[2]

See also
- Darzens reaction
- Wittig reaction
- Epoxidation
- Ylide
- E.J. Corey
- Taxol total synthesis
- Strychnine total synthesis
References
- Aggarwal, V. K.; Richardson, J. (2003). "The complexity of catalysis: origins of enantio- and diastereocontrol in sulfur ylide mediated epoxidation reactions". Chemical Communications (21): 2644–2651. doi:10.1039/b304625g. PMID 14649793.
- Aggarwal, V. K.; Winn, C. L. (2004). "Catalytic, Asymmetric Sulfur Ylide-Mediated Epoxidation of Carbonyl Compounds: Scope, Selectivity, and Applications in Synthesis". Accounts of Chemical Research. 37 (8): 611–620. doi:10.1021/ar030045f. PMID 15311960.
- Gololobov, Y. G.; Nesmeyanov, A. N.; lysenko, V. P.; Boldeskul, I. E. (1987). "Twenty-five years of dimethylsulfoxonium ethylide (corey's reagent)". Tetrahedron. 43 (12): 2609–2651. doi:10.1016/s0040-4020(01)86869-1.
- Li, A.-H.; Dai, L.-X.; Aggarwal, V. K. (1997). "Asymmetric Ylide Reactions: Epoxidation, Cyclopropanation, Aziridination, Olefination, and Rearrangement". Chemical Reviews. 97 (6): 2341–2372. doi:10.1021/cr960411r. PMID 11848902.
- Aggarwal, Varinder K.; Ford, J. Gair; Fonguerna, Sílvia; Adams, Harry; Jones, Ray V. H.; Fieldhouse, Robin (1998-08-08). "Catalytic Asymmetric Epoxidation of Aldehydes. Optimization, Mechanism, and Discovery of Stereoelectronic Control Involving a Combination of Anomeric and Cieplak Effects in Sulfur Ylide Epoxidations with Chiral 1,3-Oxathianes". Journal of the American Chemical Society. 120 (33): 8328–8339. doi:10.1021/ja9812150.
{{cite journal}}: CS1 maint: date and year (link) - McGarrigle, E. M.; Myers, E. L.; Illa, O.; Shaw, M. A.; Riches, S. L.; Aggarwal, V. K. (2007). "Chalcogenides as Organocatalysts". Chemical Reviews. 107 (12): 5841–5883. doi:10.1021/cr068402y. PMID 18072810.
- Johnson, A.W.; LaCount, R.B. (1961). "The Chemistry of Ylids. VI. Dimethylsulfonium Fluorenylide—A Synthesis of Epoxides". J. Am. Chem. Soc. 83 (2): 417–423. doi:10.1021/ja01463a040.
- Corey, E. J.; Chaykovsky, M. (1965). "Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis". J. Am. Chem. Soc. 87 (6): 1353–1364. doi:10.1021/ja01084a034.
- Li, Jack Jie (2005). Named Reactions in Heterocyclic Chemistry. Hoboken, New Jersey: John Wiley & Sons, Inc. pp. 2–14. ISBN 9780471704140.
- Mundy, Bradford, P.; Ellerd, Michael D.; Favaloro, Frank G. Jr. (2005). Name Reactions and Reagents in Organic Chemistry (2 ed.). Hoboken, New Jersey: John Wiley & Sons, Inc. pp. 174–175, 743. ISBN 9780471739869.
{{cite book}}: CS1 maint: multiple names: authors list (link) - Danishefsky, S. J.; Masters, J. J.; Young, W. B.; Link, J. T.; Snyder, L. B.; Magee, T. V.; Jung, D. K.; Isaacs, R. C. A.; Bornmann, W. G.; Alaimo, C. A.; Coburn, C. A.; Di Grandi, M. J. (1996). "Total Synthesis of Baccatin III and Taxol". Journal of the American Chemical Society. 118 (12): 2843–2859. doi:10.1021/ja952692a.
- Kuehne, M. E.; Xu, F. (1993). "Total synthesis of strychnan and aspidospermatan alkaloids. 3. The total synthesis of (.+-.)-strychnine". The Journal of Organic Chemistry. 58 (26): 7490–7497. doi:10.1021/jo00078a030.
- Luo, J.; Shea, K. J. (2010). "Polyhomologation. A Living C1 Polymerization". Accounts of Chemical Research. 43 (11): 1420–1433. doi:10.1021/ar100062a. PMID 20825177.
{kind=link}