Electrophile
In chemistry, an electrophile is a chemical species that forms bonds with nucleophiles by accepting an electron pair.[1] Because electrophiles accept electrons, they are Lewis acids.[2] Most electrophiles are positively charged, have an atom that carries a partial positive charge, or have an atom that does not have an octet of electrons.
Electrophiles mainly interact with nucleophiles through addition and substitution reactions. Frequently seen electrophiles in organic syntheses include cations such as H+ and NO+, polarized neutral molecules such as HCl, alkyl halides, acyl halides, and carbonyl compounds, polarizable neutral molecules such as Cl2 and Br2, oxidizing agents such as organic peracids, chemical species that do not satisfy the octet rule such as carbenes and radicals, and some Lewis acids such as BH3 and DIBAL.
Organic chemistry
Addition of halogens
These occur between alkenes and electrophiles, often halogens as in halogen addition reactions. Common reactions include use of bromine water to titrate against a sample to deduce the number of double bonds present. For example, ethene + bromine → 1,2-dibromoethane:
- C2H4 + Br2 → BrCH2CH2Br
This takes the form of 3 main steps shown below;[3]
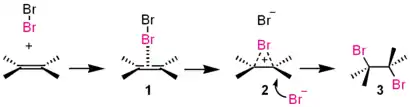
- Forming of a π-complex
- The electrophilic Br-Br molecule interacts with electron-rich alkene molecule to form a π-complex 1.
- Forming of a three-membered bromonium ion
- The alkene is working as an electron donor and bromine as an electrophile. The three-membered bromonium ion 2 consisted of two carbon atoms and a bromine atom forms with a release of Br−.
- Attacking of bromide ion
- The bromonium ion is opened by the attack of Br− from the back side. This yields the vicinal dibromide with an antiperiplanar configuration. When other nucleophiles such as water or alcohol are existing, these may attack 2 to give an alcohol or an ether.
This process is called AdE2 mechanism ("addition, electrophilic, second-order"). Iodine (I2), chlorine (Cl2), sulfenyl ion (RS+), mercury cation (Hg2+), and dichlorocarbene (:CCl2) also react through similar pathways. The direct conversion of 1 to 3 will appear when the Br− is large excess in the reaction medium. A β-bromo carbenium ion intermediate may be predominant instead of 3 if the alkene has a cation-stabilizing substituent like phenyl group. There is an example of the isolation of the bromonium ion 2.[4]
Addition of hydrogen halides
Hydrogen halides such as hydrogen chloride (HCl) adds to alkenes to give alkyl halides in hydrohalogenation. For example, the reaction of HCl with ethylene furnishes chloroethane. The reaction proceeds with a cation intermediate, being different from the above halogen addition. An example is shown below:
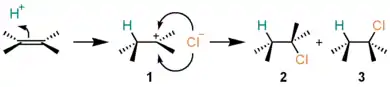
- Proton (H+) adds (by working as an electrophile) to one of the carbon atoms on the alkene to form cation 1.
- Chloride ion (Cl−) combines with the cation 1 to form the adducts 2 and 3.
In this manner, the stereoselectivity of the product, that is, from which side Cl− will attack relies on the types of alkenes applied and conditions of the reaction. At least, which of the two carbon atoms will be attacked by H+ is usually decided by Markovnikov's rule. Thus, H+ attacks the carbon atom that carries fewer substituents so as the more stabilized carbocation (with the more stabilizing substituents) will form.
This is another example of an AdE2 mechanism.[5] Hydrogen fluoride (HF) and hydrogen iodide (HI) react with alkenes in a similar manner, and Markovnikov-type products will be given. Hydrogen bromide (HBr) also takes this pathway, but sometimes a radical process competes and a mixture of isomers may form. Although introductory textbooks seldom mentions this alternative,[6] the AdE2 mechanism is generally competitive with the AdE3 mechanism (described in more detail for alkynes, below), in which transfer of the proton and nucleophilic addition occur in a concerted manner. The extent to which each pathway contributes depends on the several factors like the nature of the solvent (e.g., polarity), nucleophilicity of the halide ion, stability of the carbocation, and steric effects. As brief examples, the formation of a sterically unencumbered, stabilized carbocation favors the AdE2 pathway, while a more nucleophilic bromide ion favors the AdE3 pathway to a greater extent compared to reactions involving the chloride ion.[7]
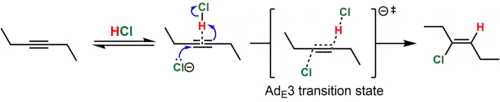
In the case of dialkyl-substituted alkynes (e.g., 3-hexyne), the intermediate vinyl cation that would result from this process is highly unstable. In such cases, the simultaneous protonation (by HCl) and attack of the alkyne by the nucleophile (Cl−) is believed to take place. This mechanistic pathway is known by the Ingold label AdE3 ("addition, electrophilic, third-order"). Because the simultaneous collision of three chemical species in a reactive orientation is improbable, the termolecular transition state is believed to be reached when the nucleophile attacks a reversibly-formed weak association of the alkyne and HCl. Such a mechanism is consistent with the predominantly anti addition (>15:1 anti:syn for the example shown) of the hydrochlorination product and the termolecular rate law, Rate = k[alkyne][HCl]2.[8][9] In support of the proposed alkyne-HCl association, a T-shaped complex of an alkyne and HCl has been characterized crystallographically.[10]
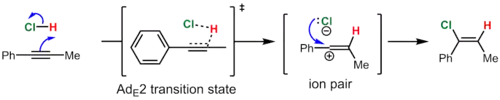
In contrast, phenylpropyne reacts by the AdE2ip ("addition, electrophilic, second-order, ion pair") mechanism to give predominantly the syn product (~10:1 syn:anti). In this case, the intermediate vinyl cation is formed by addition of HCl because it is resonance-stabilized by the phenyl group. Nevertheless, the lifetime of this high energy species is short, and the resulting vinyl cation-chloride anion ion pair immediately collapses, before the chloride ion has a chance to leave the solvent shell, to give the vinyl chloride. The proximity of the anion to the side of the vinyl cation where the proton was added is used to rationalize the observed predominance of syn addition.[7]
Hydration
One of the more complex hydration reactions utilises sulfuric acid as a catalyst. This reaction occurs in a similar way to the addition reaction but has an extra step in which the OSO3H group is replaced by an OH group, forming an alcohol:
- C2H4 + H2O → C2H5OH
As can be seen, the H2SO4 does take part in the overall reaction, however it remains unchanged so is classified as a catalyst.
This is the reaction in more detail:

- The H–OSO3H molecule has a δ+ charge on the initial H atom. This is attracted to and reacts with the double bond in the same way as before.
- The remaining (negatively charged) −OSO3H ion then attaches to the carbocation, forming ethyl hydrogensulphate (upper way on the above scheme).
- When water (H2O) is added and the mixture heated, ethanol (C2H5OH) is produced. The "spare" hydrogen atom from the water goes into "replacing" the "lost" hydrogen and, thus, reproduces sulfuric acid. Another pathway in which water molecule combines directly to the intermediate carbocation (lower way) is also possible. This pathway become predominant when aqueous sulfuric acid is used.
Overall, this process adds a molecule of water to a molecule of ethene.
This is an important reaction in industry, as it produces ethanol, whose purposes include fuels and starting material for other chemicals.
Chiral derivatives
Many electrophiles are chiral and optically stable. Typically chiral electrophiles are also optically pure.
One such reagent is the fructose-derived organocatalyst used in the Shi epoxidation.[11] The catalyst can accomplish highly enantioselective epoxidations of trans-disubstituted and trisubstituted alkenes. The Shi catalyst, a ketone, is oxidized by stoichiometric oxone to the active dioxirane form before proceeding in the catalytic cycle.
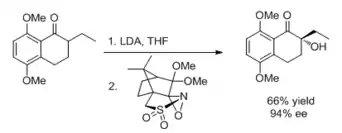
Oxaziridines such as chiral N-sulfonyloxaziridines effect enantioselective ketone alpha oxidation en route to the AB-ring segments of various natural products, including γ-rhodomycionone and α-citromycinone.[12]
Polymer-bound chiral selenium electrophiles effect asymmetric selenenylation reactions.[13] The reagents are aryl selenenyl bromides, and they were first developed for solution phase chemistry and then modified for solid phase bead attachment via an aryloxy moiety. The solid-phase reagents were applied toward the selenenylation of various alkenes with good enantioselectivities. The products can be cleaved from the solid support using organotin hydride reducing agents. Solid-supported reagents offers advantages over solution phase chemistry due to the ease of workup and purification.
Electrophilicity scale
| Fluorine | 3.86 |
| Chlorine | 3.67 |
| Bromine | 3.40 |
| Iodine | 3.09 |
| Hypochlorite | 2.52 |
| Sulfur dioxide | 2.01 |
| Carbon disulfide | 1.64 |
| Benzene | 1.45 |
| Sodium | 0.88 |
| Some selected values [14] (no dimensions) | |
Several methods exist to rank electrophiles in order of reactivity[15] and one of them is devised by Robert Parr[14] with the electrophilicity index ω given as:

with the electronegativity and chemical hardness. This equation is related to the classical equation for electrical power:



where is the resistance (Ohm or Ω) and is voltage. In this sense the electrophilicity index is a kind of electrophilic power. Correlations have been found between electrophilicity of various chemical compounds and reaction rates in biochemical systems and such phenomena as allergic contact dermititis.


An electrophilicity index also exists for free radicals.[16] Strongly electrophilic radicals such as the halogens react with electron-rich reaction sites, and strongly nucleophilic radicals such as the 2-hydroxypropyl-2-yl and tert-butyl radical react with a preference for electron-poor reaction sites.
Superelectrophiles
Superelectrophiles are defined as cationic electrophilic reagents with greatly enhanced reactivities in the presence of superacids. These compounds were first described by George A. Olah.[17] Superelectrophiles form as a doubly electron deficient superelectrophile by protosolvation of a cationic electrophile. As observed by Olah, a mixture of acetic acid and boron trifluoride is able to remove a hydride ion from isobutane when combined with hydrofluoric acid via the formation of a superacid from BF3 and HF. The responsible reactive intermediate is the [CH3CO2H3]2+ dication. Likewise, methane can be nitrated to nitromethane with nitronium tetrafluoroborate NO+
2BF−
4 only in presence of a strong acid like fluorosulfuric acid via the protonated nitronium dication.
In gitionic (gitonic) superelectrophiles, charged centers are separated by no more than one atom, for example, the protonitronium ion O=N+=O+—H (a protonated nitronium ion). And, in distonic superelectrophiles, they are separated by 2 or more atoms, for example, in the fluorination reagent F-TEDA-BF4.[18]
See also
- Nucleophile
- TRPA1,[19][20] the sensory neural target for electrophilic irritants in mammals.
References
- "Nucleophiles and Electrophiles". butane.chem.uiuc.edu. Retrieved 2020-09-21.
- "Electrophile | chemistry". Encyclopedia Britannica. Retrieved 2020-09-21.
- Lenoir, D.; Chiappe, C. (2003). "What is the Nature of the First-Formed Intermediates in the Electrophilic Halogenation of Alkenes, Alkynes, and Allenes?". Chem. Eur. J. 9 (5): 1036–1044. doi:10.1002/chem.200390097. PMID 12596140.
- Brown, R. S. (1997). "Investigation of the Early Steps in Electrophilic Bromination through the Study of the Reaction with Sterically Encumbered Olefins". Acc. Chem. Res. 30 (3): 131–137. doi:10.1021/ar960088e.
- In analogy to aromatic substitution, this process has also been termed an A-SE2 mechanism.
- Vollhardt, K. Peter C.; Schore, Neil Eric (January 2014). Organic chemistry : structure and function (7th ed.). New York, NY. ISBN 978-1-4641-2027-5. OCLC 866584251.
- H., Lowry, Thomas (1987). Mechanism and theory in organic chemistry. Richardson, Kathleen Schueller. (3rd ed.). New York: Harper & Row. ISBN 978-0060440848. OCLC 14214254.
- Fahey, Robert C.; Lee, Do-Jae. (April 1968). "Polar additions to olefins and acetylenes. V. Bimolecular and termolecular mechanisms in the hydrochlorination of acetylenes". Journal of the American Chemical Society. 90 (8): 2124–2131. doi:10.1021/ja01010a034. ISSN 0002-7863.
- A., Carroll, Felix (2010). Perspectives on structure and mechanism in organic chemistry (2nd ed.). Hoboken, N.J.: John Wiley. ISBN 9780470276105. OCLC 286483846.
- Mootz, Dietrich; Deeg, Axel (July 1992). "2-Butyne and hydrogen chloride cocrystallized: solid-state geometry of Cl-H.cntdot..cntdot..cntdot..pi. hydrogen bonding to the carbon-carbon triple bond". Journal of the American Chemical Society. 114 (14): 5887–5888. doi:10.1021/ja00040a077. ISSN 0002-7863.
- Wang, Z.; Tu, Y.; Frohn, M.; Zhang, J.; Shi, Y. (1997). "An Efficient Catalytic Asymmetric Epoxidation Method". J. Am. Chem. Soc. 119 (46): 11224–11235. doi:10.1021/ja972272g.
- Davis, F. A.; Kumar, A.; Chen, B. C. (1991). "Chemistry of oxaziridines. 16. A short, highly enantioselective synthesis of the AB-ring segments of γ-rhodomycionone and α-citromycinone using (+)-[(8,8-dimethoxycamphoryl)sulfonyl]oxaziridine". J. Org. Chem. 56 (3): 1143–1145. doi:10.1021/jo00003a042.
- Uehlin, L.; Wirth, T. (2001). "Novel Polymer-Bound Chiral Selenium Electrophiles". Org. Lett. 3 (18): 2931–2933. doi:10.1021/ol0164435. PMID 11529793.
- Parr, R. G.; Szentpaly, L. v.; Liu, S. (1999). "Electrophilicity Index". J. Am. Chem. Soc. 121 (9): 1922–1924. doi:10.1021/ja983494x.
- Chattaraj, P. K.; Sarkar, U.; Roy, D. R. (2006). "Electrophilicity Index". Chem. Rev. 106 (6): 2065–2091. doi:10.1021/cr040109f. PMID 16771443.
- De Vleeschouwer, Freija; Speybroeck, Veronique Van; Waroquier, Michel; Geerlings, Paul; De Proft, Frank (2007). "Electrophilicity and Nucleophilicity Index for Radicals". Org. Lett. 9 (14): 2721–2724. doi:10.1021/ol071038k. PMID 17559221.
- Olah, George A.; Germain, Alain; Lin, Henry C.; Forsyth, David A. (1975). "Electrophilic reactions at single bonds. XVIII. Indication of protosolvated de facto substituting agents in the reactions of alkanes with acetylium and nitronium ions in superacidic media". J. Am. Chem. Soc. 97 (10): 2928–2929. doi:10.1021/ja00843a067.
- Solingapuram Sai, Kiran Kumar; Gilbert, Thomas M.; Klumpp, Douglas A. (2007). "Knorr Cyclizations and Distonic Superelectrophiles". J. Org. Chem. 72 (25): 9761–9764. doi:10.1021/jo7013092. PMID 17999519.
- Lin King, John V.; Emrick, Joshua J.; Kelly, Mark J. S.; Herzig, Volker; King, Glenn F.; Medzihradszky, Katalin F.; Julius, David (5 September 2019). "A Cell-Penetrating Scorpion Toxin Enables Mode-Specific Modulation of TRPA1 and Pain". Cell. 178 (6): 1362–1374.e16. doi:10.1016/j.cell.2019.07.014. ISSN 1097-4172. PMC 6731142. PMID 31447178.
- Zhao, Jianhua; Lin King, John V.; Paulsen, Candice E.; Cheng, Yifan; Julius, David (2020-07-08). "Irritant-evoked activation and calcium modulation of the TRPA1 receptor". Nature. 585 (7823): 141–145. doi:10.1038/s41586-020-2480-9. ISSN 1476-4687. PMC 7483980. PMID 32641835.