Motor neuron disease
Motor neuron diseases or motor neurone diseases (MNDs) are a group of rare neurodegenerative disorders that selectively affect motor neurons, the cells which control voluntary muscles of the body.[1][2] They include amyotrophic lateral sclerosis (ALS),[3][4] progressive bulbar palsy (PBP), pseudobulbar palsy, progressive muscular atrophy (PMA), primary lateral sclerosis (PLS), spinal muscular atrophy (SMA) and monomelic amyotrophy (MMA), as well as some rarer variants resembling ALS.
| Motor neuron disease | |
|---|---|
 | |
| spinal diagram | |
| Specialty | Neurology |
Motor neuron diseases affect both children and adults.[5] While each motor neuron disease affects patients differently, they all cause movement-related symptoms, mainly muscle weakness.[6] Most of these diseases seem to occur randomly without known causes, but some forms are inherited.[2] Studies into these inherited forms have led to discoveries of various genes (e.g. SOD1) that are thought to be important in understanding how the disease occurs.[7]
Symptoms of motor neuron diseases can be first seen at birth or can come on slowly later in life. Most of these diseases worsen over time; while some, such as ALS, shorten one's life expectancy, others do not.[2] Currently, there are no approved treatments for the majority of motor neuron disorders, and care is mostly symptomatic.[2]
Signs and symptoms

Signs and symptoms depend on the specific disease, but motor neuron diseases typically manifest as a group of movement-related symptoms.[6] They come on slowly, and worsen over the course of more than three months. Various patterns of muscle weakness are seen, and muscle cramps and spasms may occur. One can have difficulty breathing with climbing stairs (exertion), difficulty breathing when lying down (orthopnea), or even respiratory failure if breathing muscles become involved. Bulbar symptoms, including difficulty speaking (dysarthria), difficulty swallowing (dysphagia), and excessive saliva production (sialorrhea), can also occur. Sensation, or the ability to feel, is typically not affected. Emotional disturbance (e.g. pseudobulbar affect) and cognitive and behavioural changes (e.g. problems in word fluency, decision-making, and memory) are also seen.[2][6] There can be lower motor neuron findings (e.g. muscle wasting, muscle twitching), upper motor neuron findings (e.g. brisk reflexes, Babinski reflex, Hoffman's reflex, increased muscle tone), or both.[6]
Motor neuron diseases are seen both in children and adults.[2] Those that affect children tend to be inherited or familial, and their symptoms are either present at birth or appear before learning to walk. Those that affect adults tend to appear after age 40.[2] The clinical course depends on the specific disease, but most progress or worsen over the course of months.[6] Some are fatal (e.g. ALS), while others are not (e.g. PLS).[2]
Patterns of weakness
Various patterns of muscle weakness occur in different motor neuron diseases.[6] Weakness can be symmetric or asymmetric, and it can occur in body parts that are distal, proximal, or both. According to Statland et al., there are three main weakness patterns that are seen in motor neuron diseases, which are:[6][9]
- Asymmetric distal weakness without sensory loss (e.g. ALS, PLS, PMA, MMA)
- Symmetric weakness without sensory loss (e.g. PMA, PLS)
- Symmetric focal midline proximal weakness (neck, trunk, bulbar involvement; e.g. ALS, PBP, PLS)
Lower and upper motor neuron findings
Motor neuron diseases are on a spectrum in terms of upper and lower motor neuron involvement.[6] Some have just lower or upper motor neuron findings, while others have a mix of both. Lower motor neuron (LMN) findings include muscle atrophy and fasciculations, and upper motor neuron (UMN) findings include hyperreflexia, spasticity, muscle spasm, and abnormal reflexes.[2][6]
Pure upper motor neuron diseases, or those with just UMN findings, include PLS.[10]
Pure lower motor neuron diseases, or those with just LMN findings, include PMA.[11]
Motor neuron diseases with both UMN and LMN findings include both familial and sporadic ALS.[12]
Causes
Most cases are sporadic and their causes are usually not known.[2] It is thought that environmental, toxic, viral, or genetic factors may be involved.[2]
DNA damage
TARDBP (TAR DNA-binding protein 43), also referred to as TDP-43, is a critical component of the non-homologous end joining (NHEJ) enzymatic pathway that repairs DNA double-strand breaks in pluripotent stem cell-derived motor neurons.[13] TDP-43 is rapidly recruited to double-strand breaks where it acts as a scaffold for the recruitment of the XRCC4-DNA ligase protein complex that then acts to repair double-strand breaks. About 95% of ALS patients have abnormalities in the nucleus-cytoplasmic localization in spinal motor neurons of TDP43. In TDP-43 depleted human neural stem cell-derived motor neurons, as well as in sporadic ALS patients’ spinal cord specimens there is significant double-strand break accumulation and reduced levels of NHEJ.[13]
Associated risk factors
In adults, men are more commonly affected than women.[2]
Diagnosis
Differential diagnosis can be challenging due to the number of overlapping symptoms, shared between several motor neuron diseases.[14] Frequently, the diagnosis is based on clinical findings (i.e. LMN vs. UMN signs and symptoms, patterns of weakness), family history of MND, and a variation of tests, many of which are used to rule out disease mimics, which can manifest with identical symptoms.[15][16]
Classification
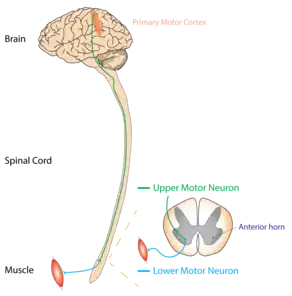
Motor neuron disease describes a collection of clinical disorders, characterized by progressive muscle weakness and the degeneration of the motor neuron on electrophysiological testing. The term "motor neuron disease" has varying meanings in different countries. Similarly, the literature inconsistently classifies which degenerative motor neuron disorders can be included under the umbrella term "motor neuron disease". The four main types of MND are marked (*) in the table below.[17]
All types of MND can be differentiated by two defining characteristics:[6]
- Is the disease sporadic or inherited?
- Is there involvement of the upper motor neurons (UMN), the lower motor neurons (LMN), or both?
Sporadic or acquired MNDs occur in patients with no family history of degenerative motor neuron disease. Inherited or genetic MNDs adhere to one of the following inheritance patterns: autosomal dominant, autosomal recessive, or X-linked. Some disorders, like ALS, can occur sporadically (85%) or can have a genetic cause (15%) with the same clinical symptoms and progression of disease.[6]
UMNs are motor neurons that project from the cortex down to the brainstem or spinal cord.[18] LMNs originate in the anterior horns of the spinal cord and synapse on peripheral muscles.[18] Both motor neurons are necessary for the strong contraction of a muscle, but damage to an UMN can be distinguished from damage to a LMN by physical exam.[19]
| Type | UMN degeneration | LMN degeneration |
|---|---|---|
| Sporadic MNDs | ||
| Sporadic amyotrophic lateral sclerosis (ALS)* | Yes[6] | Yes[6] |
| Primary lateral sclerosis (PLS)* | Yes[6] | No[6] |
| Progressive muscular atrophy (PMA)* | No[6] | Yes[6] |
| Progressive bulbar palsy (PBP)* | Yes[17] | Yes, bulbar region[17] |
| Pseudobulbar palsy | Yes, bulbar region[6] | No[6] |
| Monomelic amyotrophy (MMA) | No | Yes |
| Inherited MNDs | ||
| Familial amyotrophic lateral sclerosis (ALS)* | Yes[6] | Yes[6] |
Tests
- Cerebrospinal fluid (CSF) tests: Analysis of the fluid from around the brain and spinal cord could reveal signs of an infection or inflammation.[20]
- Magnetic resonance imaging (MRI): An MRI of the brain and spinal cord is recommended in patients with UMN signs and symptoms to explore other causes, such as a tumor, inflammation, or lack of blood supply (stroke).[20]
- Electromyogram (EMG) & nerve conduction study (NCS): The EMG, which evaluates muscle function, and NCS, which evaluates nerve function, are performed together in patients with LMN signs.
- For patients with MND affecting the LMNs, the EMG will show evidence of: (1) acute denervation, which is ongoing as motor neurons degenerate, and (2) chronic denervation and reinnervation of the muscle, as the remaining motor neurons attempt to fill in for lost motor neurons.[20]
- By contrast, the NCS in these patients is usually normal. It can show a low compound muscle action potential (CMAP), which results from the loss of motor neurons, but the sensory neurons should remain unaffected.[21]
- Tissue biopsy: Taking a small sample of a muscle or nerve may be necessary if the EMG/NCS is not specific enough to rule out other causes of progressive muscle weakness, but it is rarely used.
Treatment
There are no known curative treatments for the majority of motor neuron disorders. Please refer to the articles on individual disorders for more details.[22]
Prognosis
The table below lists life expectancy for patients who are diagnosed with MND.
| Type | Median survival time from start of symptoms |
|---|---|
| Amyotrophic lateral sclerosis (ALS) | 2–5 years[20][23] |
| Primary lateral sclerosis (PLS) | 8–10 years[20] |
| Progressive muscular atrophy (PMA) | 2–4 years[20] |
| Progressive bulbar palsy (PBP) | 6 months – 3 years[23] |
| Pseudobulbar palsy | No change in survival |
Terminology
In the United States and Canada, the term motor neuron disease usually refers to the group of disorders while amyotrophic lateral sclerosis is frequently called Lou Gehrig's disease.[2][5][24] In the United Kingdom and Australia, the term motor neuron(e) disease is used for amyotrophic lateral sclerosis,[3][4] although is not uncommon to refer to the entire group.[25][26]
While MND refers to a specific subset of similar diseases, there are numerous other diseases of motor neurons that are referred to collectively as "motor neuron disorders", for instance the diseases belonging to the spinal muscular atrophies group.[1] However, they are not classified as "motor neuron diseases" by the 11th edition of the International Statistical Classification of Diseases and Related Health Problems (ICD-11),[27] which is the definition followed in this article.
See also
- Spinal muscular atrophies
- Hereditary motor and sensory neuropathies
References
- Ince PG, Clark B, Holton J, Revesz T, Wharton SB (2008). "Chapter 13: Diseases of movement and system degenerations". In Greenfield JG, Love S, Louis DN, Ellison DW (eds.). Greenfield's neuropathology. Vol. 1 (8th ed.). London: Hodder Arnold. p. 947. ISBN 978-0-340-90681-1.
- "Motor Neuron Diseases Fact Sheet: National Institute of Neurological Disorders and Stroke (NINDS)". www.ninds.nih.gov. Archived from the original on 13 April 2014. Retrieved 7 November 2010.
- "Motor neurone disease – NHS". nhs.uk. 15 January 2018. Retrieved 24 October 2020.
- Healthdirect Australia (17 April 2020). "Motor neurone disease (MND)". www.healthdirect.gov.au. Retrieved 24 October 2020.
- Cooper-Knock J, Jenkins T, Shaw PJ (1 September 2013). Clinical and molecular aspects of motor neuron disease. San Rafael, California. ISBN 978-1-61504-429-0. OCLC 860981760.
- Statland JM, Barohn RJ, McVey AL, Katz JS, Dimachkie MM (November 2015). "Patterns of Weakness, Classification of Motor Neuron Disease, and Clinical Diagnosis of Sporadic Amyotrophic Lateral Sclerosis". Neurologic Clinics. 33 (4): 735–748. doi:10.1016/j.ncl.2015.07.006. PMC 4629510. PMID 26515618.
- Cooper-Knock J, Jenkins T, Shaw PJ (1 September 2013). Clinical and molecular aspects of motor neuron disease. San Rafael, California (1537 Fourth Street, San Rafael, CA 94901 USA). ISBN 9781615044290. OCLC 860981760.
{{cite book}}: CS1 maint: location (link) - "Patient with amyotrophic lateral sclerosis (ALS) (case | Open-i". openi.nlm.nih.gov. Archived from the original on 15 December 2018. Retrieved 12 December 2018.
- Barohn RJ, Amato AA (May 2013). "Pattern-recognition approach to neuropathy and neuronopathy". Neurologic Clinics. 31 (2): 343–361. doi:10.1016/j.ncl.2013.02.001. PMC 3922643. PMID 23642713.
- Emos MC, Agarwal S (2022). "Neuroanatomy, Upper Motor Neuron Lesion". StatPearls. Treasure Island (FL): StatPearls Publishing. PMID 30725990. Retrieved 24 June 2022.
- "Progressive Muscular Atrophy - an overview | ScienceDirect Topics". www.sciencedirect.com. Retrieved 24 June 2022.
- "Motor Neuron Diseases Fact Sheet | National Institute of Neurological Disorders and Stroke". www.ninds.nih.gov. Retrieved 24 June 2022.
- Mitra J, Guerrero EN, Hegde PM, Liachko NF, Wang H, Vasquez V, et al. (March 2019). "Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects". Proceedings of the National Academy of Sciences of the United States of America. 116 (10): 4696–4705. doi:10.1073/pnas.1818415116. PMC 6410842. PMID 30770445.
- Statland, Jeffrey M.; Barohn, Richard J.; McVey, April L.; Katz, Jonathan; Dimachkie, Mazen M. (2015). "Patterns of Weakness, Classification of Motor Neuron Disease & Clinical Diagnosis of Sporadic ALS". Neurologic Clinics. 33 (4): 735–748. doi:10.1016/j.ncl.2015.07.006. ISSN 0733-8619. PMC 4629510. PMID 26515618.
- "Archive | Practical Neurology". pn.bmj.com. Retrieved 24 June 2022.
- Foster LA, Salajegheh MK (January 2019). "Motor Neuron Disease: Pathophysiology, Diagnosis, and Management". The American Journal of Medicine. 132 (1): 32–37. doi:10.1016/j.amjmed.2018.07.012. PMID 30075105. S2CID 51910723.
- "What forms does MND take?". www.mndnsw.asn.au. Retrieved 11 December 2018.
- Blumenfeld H (2002). Neuroanatomy through clinical cases. Sunderland, Mass.: Sinauer. ISBN 087893060-4. OCLC 44628054.
- Javed K, Daly DT (2022). "Neuroanatomy, Lower Motor Neuron Lesion". StatPearls. Treasure Island (FL): StatPearls Publishing. PMID 30969636. Retrieved 24 June 2022.
- Foster LA, Salajegheh MK (January 2019). "Motor Neuron Disease: Pathophysiology, Diagnosis, and Management". The American Journal of Medicine. 132 (1): 32–37. doi:10.1016/j.amjmed.2018.07.012. PMID 30075105. S2CID 51910723.
- Duleep A, Shefner J (February 2013). "Electrodiagnosis of motor neuron disease". Physical Medicine and Rehabilitation Clinics of North America. 24 (1): 139–151. doi:10.1016/j.pmr.2012.08.022. PMID 23177036.
- "NIH: ninds: Motor Neuron Diseases Information Page". 27 March 2019. Retrieved 18 November 2019.
- "Different types of MND". Irish Motor Neurone Disease Association. Retrieved 12 December 2018.
- Shaw PJ (August 2005). "Molecular and cellular pathways of neurodegeneration in motor neurone disease". Journal of Neurology, Neurosurgery, and Psychiatry. 76 (8): 1046–1057. doi:10.1136/jnnp.2004.048652. PMC 1739758. PMID 16024877.
Many doctors use the terms motor neuron disease and ALS interchangeably.
- "An introduction to motor neurone disease (MND)" (PDF). motor neurone disease association. 2015. Archived (PDF) from the original on 9 October 2022.
- Schapira AH, Wszolek ZK, Dawson TM, Wood NW (13 February 2017). Neurodegeneration. Chichester, West Sussex. ISBN 978-1-118-66191-8. OCLC 958876527.
- "8B60 Motor neuron disease". ICD-11 for Mortality and Morbidity Statistics. World Health Organisation.
External links
 Media related to Motor neuron diseases at Wikimedia Commons
Media related to Motor neuron diseases at Wikimedia Commons- Motor neuron diseases at NINDS