Transcription (biology)
Transcription is the process of copying a segment of DNA into RNA. The segments of DNA transcribed into RNA molecules that can encode proteins are said to produce messenger RNA (mRNA). Other segments of DNA are copied into RNA molecules called non-coding RNAs (ncRNAs). mRNA comprises only 1-3% of total RNA samples.[1] Less than 2% of the human genome can be transcribed into mRNA (Human genome#Coding vs. noncoding DNA), while at least 80% of mammalian genomic DNA can be actively transcribed (in one or more types of cells), with the majority of this 80% considered to be ncRNA.[2]

Both DNA and RNA are nucleic acids, which use base pairs of nucleotides as a complementary language. During transcription, a DNA sequence is read by an RNA polymerase, which produces a complementary, antiparallel RNA strand called a primary transcript.
Transcription proceeds in the following general steps:
- RNA polymerase, together with one or more general transcription factors, binds to promoter DNA.
- RNA polymerase generates a transcription bubble, which separates the two strands of the DNA helix. This is done by breaking the hydrogen bonds between complementary DNA nucleotides.
- RNA polymerase adds RNA nucleotides (which are complementary to the nucleotides of one DNA strand).
- RNA sugar-phosphate backbone forms with assistance from RNA polymerase to form an RNA strand.
- Hydrogen bonds of the RNA–DNA helix break, freeing the newly synthesized RNA strand.
- If the cell has a nucleus, the RNA may be further processed. This may include polyadenylation, capping, and splicing.
- The RNA may remain in the nucleus or exit to the cytoplasm through the nuclear pore complex.
If the stretch of DNA is transcribed into an RNA molecule that encodes a protein, the RNA is termed messenger RNA (mRNA); the mRNA, in turn, serves as a template for the protein's synthesis through translation. Other stretches of DNA may be transcribed into small non-coding RNAs such as microRNA, transfer RNA (tRNA), small nucleolar RNA (snoRNA), small nuclear RNA (snRNA), or enzymatic RNA molecules called ribozymes[3] as well as larger non-coding RNAs such as ribosomal RNA (rRNA), and long non-coding RNA (lncRNA). Overall, RNA helps synthesize, regulate, and process proteins; it therefore plays a fundamental role in performing functions within a cell.
In virology, the term transcription may also be used when referring to mRNA synthesis from an RNA molecule (i.e., equivalent to RNA replication). For instance, the genome of a negative-sense single-stranded RNA (ssRNA -) virus may be a template for a positive-sense single-stranded RNA (ssRNA +). This is because the positive-sense strand contains the sequence information needed to translate the viral proteins needed for viral replication. This process is catalyzed by a viral RNA replicase.[4]
Background
A DNA transcription unit encoding for a protein may contain both a coding sequence, which will be translated into the protein, and regulatory sequences, which direct and regulate the synthesis of that protein. The regulatory sequence before ("upstream" from) the coding sequence is called the five prime untranslated region (5'UTR); the sequence after ("downstream" from) the coding sequence is called the three prime untranslated region (3'UTR).[3]
As opposed to DNA replication, transcription results in an RNA complement that includes the nucleotide uracil (U) in all instances where thymine (T) would have occurred in a DNA complement.
Only one of the two DNA strands serve as a template for transcription. The antisense strand of DNA is read by RNA polymerase from the 3' end to the 5' end during transcription (3' → 5'). The complementary RNA is created in the opposite direction, in the 5' → 3' direction, matching the sequence of the sense strand with the exception of switching uracil for thymine. This directionality is because RNA polymerase can only add nucleotides to the 3' end of the growing mRNA chain. This use of only the 3' → 5' DNA strand eliminates the need for the Okazaki fragments that are seen in DNA replication.[3] This also removes the need for an RNA primer to initiate RNA synthesis, as is the case in DNA replication.
The non-template (sense) strand of DNA is called the coding strand, because its sequence is the same as the newly created RNA transcript (except for the substitution of uracil for thymine). This is the strand that is used by convention when presenting a DNA sequence.[5]
Transcription has some proofreading mechanisms, but they are fewer and less effective than the controls for copying DNA. As a result, transcription has a lower copying fidelity than DNA replication.[6]
Major steps
Transcription is divided into initiation, promoter escape, elongation, and termination.[7]
Setting up for transcription
Enhancers, transcription factors, Mediator complex and DNA loops in mammalian transcription
Setting up for transcription in mammals is regulated by many cis-regulatory elements, including core promoter and promoter-proximal elements that are located near the transcription start sites of genes. Core promoters combined with general transcription factors are sufficient to direct transcription initiation, but generally have low basal activity.[8] Other important cis-regulatory modules are localized in DNA regions that are distant from the transcription start sites. These include enhancers, silencers, insulators and tethering elements.[9] Among this constellation of elements, enhancers and their associated transcription factors have a leading role in the initiation of gene transcription.[10] An enhancer localized in a DNA region distant from the promoter of a gene can have a very large effect on gene transcription, with some genes undergoing up to 100-fold increased transcription due to an activated enhancer.[11]
Enhancers are regions of the genome that are major gene-regulatory elements. Enhancers control cell-type-specific gene transcription programs, most often by looping through long distances to come in physical proximity with the promoters of their target genes.[12] While there are hundreds of thousands of enhancer DNA regions,[13] for a particular type of tissue only specific enhancers are brought into proximity with the promoters that they regulate. In a study of brain cortical neurons, 24,937 loops were found, bringing enhancers to their target promoters.[11] Multiple enhancers, each often at tens or hundred of thousands of nucleotides distant from their target genes, loop to their target gene promoters and can coordinate with each other to control transcription of their common target gene.[12]
The schematic illustration in this section shows an enhancer looping around to come into close physical proximity with the promoter of a target gene. The loop is stabilized by a dimer of a connector protein (e.g. dimer of CTCF or YY1), with one member of the dimer anchored to its binding motif on the enhancer and the other member anchored to its binding motif on the promoter (represented by the red zigzags in the illustration).[14] Several cell function specific transcription factors (there are about 1,600 transcription factors in a human cell[15]) generally bind to specific motifs on an enhancer[16] and a small combination of these enhancer-bound transcription factors, when brought close to a promoter by a DNA loop, govern level of transcription of the target gene. Mediator (a complex usually consisting of about 26 proteins in an interacting structure) communicates regulatory signals from enhancer DNA-bound transcription factors directly to the RNA polymerase II (pol II) enzyme bound to the promoter.[17]
Enhancers, when active, are generally transcribed from both strands of DNA with RNA polymerases acting in two different directions, producing two enhancer RNAs (eRNAs) as illustrated in the Figure.[18] An inactive enhancer may be bound by an inactive transcription factor. Phosphorylation of the transcription factor may activate it and that activated transcription factor may then activate the enhancer to which it is bound (see small red star representing phosphorylation of transcription factor bound to enhancer in the illustration).[19] An activated enhancer begins transcription of its RNA before activating transcription of messenger RNA from its target gene.[20]
CpG island methylation and demethylation

Transcription regulation at about 60% of promoters is also controlled by methylation of cytosines within CpG dinucleotides (where 5’ cytosine is followed by 3’ guanine or CpG sites). 5-methylcytosine (5-mC) is a methylated form of the DNA base cytosine (see Figure). 5-mC is an epigenetic marker found predominantly within CpG sites. About 28 million CpG dinucleotides occur in the human genome.[21] In most tissues of mammals, on average, 70% to 80% of CpG cytosines are methylated (forming 5-methylCpG or 5-mCpG).[22] Methylated cytosines within 5’cytosine-guanine 3’ sequences often occur in groups, called CpG islands. About 60% of promoter sequences have a CpG island while only about 6% of enhancer sequences have a CpG island.[23] CpG islands constitute regulatory sequences, since if CpG islands are methylated in the promoter of a gene this can reduce or silence gene transcription.[24]
DNA methylation regulates gene transcription through interaction with methyl binding domain (MBD) proteins, such as MeCP2, MBD1 and MBD2. These MBD proteins bind most strongly to highly methylated CpG islands.[25] These MBD proteins have both a methyl-CpG-binding domain as well as a transcription repression domain.[25] They bind to methylated DNA and guide or direct protein complexes with chromatin remodeling and/or histone modifying activity to methylated CpG islands. MBD proteins generally repress local chromatin such as by catalyzing the introduction of repressive histone marks, or creating an overall repressive chromatin environment through nucleosome remodeling and chromatin reorganization.[25]
As noted in the previous section, transcription factors are proteins that bind to specific DNA sequences in order to regulate the expression of a gene. The binding sequence for a transcription factor in DNA is usually about 10 or 11 nucleotides long. As summarized in 2009, Vaquerizas et al. indicated there are approximately 1,400 different transcription factors encoded in the human genome by genes that constitute about 6% of all human protein encoding genes.[26] About 94% of transcription factor binding sites (TFBSs) that are associated with signal-responsive genes occur in enhancers while only about 6% of such TFBSs occur in promoters.[16]
EGR1 protein is a particular transcription factor that is important for regulation of methylation of CpG islands. An EGR1 transcription factor binding site is frequently located in enhancer or promoter sequences.[27] There are about 12,000 binding sites for EGR1 in the mammalian genome and about half of EGR1 binding sites are located in promoters and half in enhancers.[27] The binding of EGR1 to its target DNA binding site is insensitive to cytosine methylation in the DNA.[27]
While only small amounts of EGR1 transcription factor protein are detectable in cells that are un-stimulated, translation of the EGR1 gene into protein at one hour after stimulation is drastically elevated.[28] Production of EGR1 transcription factor proteins, in various types of cells, can be stimulated by growth factors, neurotransmitters, hormones, stress and injury.[28] In the brain, when neurons are activated, EGR1 proteins are up-regulated and they bind to (recruit) the pre-existing TET1 enzymes that are produced in high amounts in neurons. TET enzymes can catalyse demethylation of 5-methylcytosine. When EGR1 transcription factors bring TET1 enzymes to EGR1 binding sites in promoters, the TET enzymes can demethylate the methylated CpG islands at those promoters. Upon demethylation, these promoters can then initiate transcription of their target genes. Hundreds of genes in neurons are differentially expressed after neuron activation through EGR1 recruitment of TET1 to methylated regulatory sequences in their promoters.[27]
The methylation of promoters is also altered in response to signals. The three mammalian DNA methyltransferasess (DNMT1, DNMT3A, and DNMT3B) catalyze the addition of methyl groups to cytosines in DNA. While DNMT1 is a “maintenance” methyltransferase, DNMT3A and DNMT3B can carry out new methylations. There are also two splice protein isoforms produced from the DNMT3A gene: DNA methyltransferase proteins DNMT3A1 and DNMT3A2.[29]
The splice isoform DNMT3A2 behaves like the product of a classical immediate-early gene and, for instance, it is robustly and transiently produced after neuronal activation.[30] Where the DNA methyltransferase isoform DNMT3A2 binds and adds methyl groups to cytosines appears to be determined by histone post translational modifications.[31][32][33]
On the other hand, neural activation causes degradation of DNMT3A1 accompanied by reduced methylation of at least one evaluated targeted promoter.[34]
Initiation
Transcription begins with the binding of RNA polymerase, together with one or more general transcription factors, to a specific DNA sequence referred to as a "promoter" to form an RNA polymerase-promoter "closed complex". In the "closed complex" the promoter DNA is still fully double-stranded.[7]
RNA polymerase, assisted by one or more general transcription factors, then unwinds approximately 14 base pairs of DNA to form an RNA polymerase-promoter "open complex". In the "open complex" the promoter DNA is partly unwound and single-stranded. The exposed, single-stranded DNA is referred to as the "transcription bubble."[7]
RNA polymerase, assisted by one or more general transcription factors, then selects a transcription start site in the transcription bubble, binds to an initiating NTP and an extending NTP (or a short RNA primer and an extending NTP) complementary to the transcription start site sequence, and catalyzes bond formation to yield an initial RNA product.[7]
In bacteria, RNA polymerase holoenzyme consists of five subunits: 2 α subunits, 1 β subunit, 1 β' subunit, and 1 ω subunit. In bacteria, there is one general RNA transcription factor known as a sigma factor. RNA polymerase core enzyme binds to the bacterial general transcription (sigma) factor to form RNA polymerase holoenzyme and then binds to a promoter.[7] (RNA polymerase is called a holoenzyme when sigma subunit is attached to the core enzyme which is consist of 2 α subunits, 1 β subunit, 1 β' subunit only). Unlike eukaryotes, the initiating nucleotide of nascent bacterial mRNA is not capped with a modified guanine nucleotide. The initiating nucleotide of bacterial transcripts bears a 5′ triphosphate (5′-PPP), which can be used for genome-wide mapping of transcription initiation sites.[35]
In archaea and eukaryotes, RNA polymerase contains subunits homologous to each of the five RNA polymerase subunits in bacteria and also contains additional subunits. In archaea and eukaryotes, the functions of the bacterial general transcription factor sigma are performed by multiple general transcription factors that work together.[7] In archaea, there are three general transcription factors: TBP, TFB, and TFE. In eukaryotes, in RNA polymerase II-dependent transcription, there are six general transcription factors: TFIIA, TFIIB (an ortholog of archaeal TFB), TFIID (a multisubunit factor in which the key subunit, TBP, is an ortholog of archaeal TBP), TFIIE (an ortholog of archaeal TFE), TFIIF, and TFIIH. The TFIID is the first component to bind to DNA due to binding of TBP, while TFIIH is the last component to be recruited. In archaea and eukaryotes, the RNA polymerase-promoter closed complex is usually referred to as the "preinitiation complex."[36]
Transcription initiation is regulated by additional proteins, known as activators and repressors, and, in some cases, associated coactivators or corepressors, which modulate formation and function of the transcription initiation complex.[7]
Promoter escape
After the first bond is synthesized, the RNA polymerase must escape the promoter. During this time there is a tendency to release the RNA transcript and produce truncated transcripts. This is called abortive initiation, and is common for both eukaryotes and prokaryotes.[37] Abortive initiation continues to occur until an RNA product of a threshold length of approximately 10 nucleotides is synthesized, at which point promoter escape occurs and a transcription elongation complex is formed.
Mechanistically, promoter escape occurs through DNA scrunching, providing the energy needed to break interactions between RNA polymerase holoenzyme and the promoter.[38]
In bacteria, it was historically thought that the sigma factor is definitely released after promoter clearance occurs. This theory had been known as the obligate release model. However, later data showed that upon and following promoter clearance, the sigma factor is released according to a stochastic model known as the stochastic release model.[39]
In eukaryotes, at an RNA polymerase II-dependent promoter, upon promoter clearance, TFIIH phosphorylates serine 5 on the carboxy terminal domain of RNA polymerase II, leading to the recruitment of capping enzyme (CE).[40][41] The exact mechanism of how CE induces promoter clearance in eukaryotes is not yet known.
Elongation
One strand of the DNA, the template strand (or noncoding strand), is used as a template for RNA synthesis. As transcription proceeds, RNA polymerase traverses the template strand and uses base pairing complementarity with the DNA template to create an RNA copy (which elongates during the traversal). Although RNA polymerase traverses the template strand from 3' → 5', the coding (non-template) strand and newly formed RNA can also be used as reference points, so transcription can be described as occurring 5' → 3'. This produces an RNA molecule from 5' → 3', an exact copy of the coding strand (except that thymines are replaced with uracils, and the nucleotides are composed of a ribose (5-carbon) sugar where DNA has deoxyribose (one fewer oxygen atom) in its sugar-phosphate backbone).
mRNA transcription can involve multiple RNA polymerases on a single DNA template and multiple rounds of transcription (amplification of particular mRNA), so many mRNA molecules can be rapidly produced from a single copy of a gene. The characteristic elongation rates in prokaryotes and eukaryotes are about 10-100 nts/sec.[42] In eukaryotes, however, nucleosomes act as major barriers to transcribing polymerases during transcription elongation.[43][44] In these organisms, the pausing induced by nucleosomes can be regulated by transcription elongation factors such as TFIIS.[44]
Elongation also involves a proofreading mechanism that can replace incorrectly incorporated bases. In eukaryotes, this may correspond with short pauses during transcription that allow appropriate RNA editing factors to bind. These pauses may be intrinsic to the RNA polymerase or due to chromatin structure.
Termination
Bacteria use two different strategies for transcription termination – Rho-independent termination and Rho-dependent termination. In Rho-independent transcription termination, RNA transcription stops when the newly synthesized RNA molecule forms a G-C-rich hairpin loop followed by a run of Us. When the hairpin forms, the mechanical stress breaks the weak rU-dA bonds, now filling the DNA–RNA hybrid. This pulls the poly-U transcript out of the active site of the RNA polymerase, terminating transcription. In the "Rho-dependent" type of termination, a protein factor called "Rho" destabilizes the interaction between the template and the mRNA, thus releasing the newly synthesized mRNA from the elongation complex.[45]
Transcription termination in eukaryotes is less well understood than in bacteria, but involves cleavage of the new transcript followed by template-independent addition of adenines at its new 3' end, in a process called polyadenylation.[46]
Role of RNA polymerase in post-transcriptional changes in RNA
RNA polymerase plays a very crucial role in all steps including post-transcriptional changes in RNA.
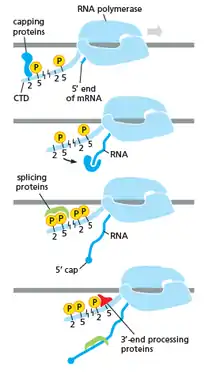
As shown in the image in the right it is evident that the CTD (C Terminal Domain) is a tail that changes its shape; this tail will be used as a carrier of splicing, capping and polyadenylation, as shown in the image on the left.[47]
Inhibitors
Transcription inhibitors can be used as antibiotics against, for example, pathogenic bacteria (antibacterials) and fungi (antifungals). An example of such an antibacterial is rifampicin, which inhibits bacterial transcription of DNA into mRNA by inhibiting DNA-dependent RNA polymerase by binding its beta-subunit, while 8-hydroxyquinoline is an antifungal transcription inhibitor.[48] The effects of histone methylation may also work to inhibit the action of transcription. Potent, bioactive natural products like triptolide that inhibit mammalian transcription via inhibition of the XPB subunit of the general transcription factor TFIIH has been recently reported as a glucose conjugate for targeting hypoxic cancer cells with increased glucose transporter production.[49]
Endogenous inhibitors
In vertebrates, the majority of gene promoters contain a CpG island with numerous CpG sites.[50] When many of a gene's promoter CpG sites are methylated the gene becomes inhibited (silenced).[51] Colorectal cancers typically have 3 to 6 driver mutations and 33 to 66 hitchhiker or passenger mutations.[52] However, transcriptional inhibition (silencing) may be of more importance than mutation in causing progression to cancer. For example, in colorectal cancers about 600 to 800 genes are transcriptionally inhibited by CpG island methylation (see regulation of transcription in cancer). Transcriptional repression in cancer can also occur by other epigenetic mechanisms, such as altered production of microRNAs.[53] In breast cancer, transcriptional repression of BRCA1 may occur more frequently by over-produced microRNA-182 than by hypermethylation of the BRCA1 promoter (see Low expression of BRCA1 in breast and ovarian cancers).
Transcription factories
Active transcription units are clustered in the nucleus, in discrete sites called transcription factories or euchromatin. Such sites can be visualized by allowing engaged polymerases to extend their transcripts in tagged precursors (Br-UTP or Br-U) and immuno-labeling the tagged nascent RNA. Transcription factories can also be localized using fluorescence in situ hybridization or marked by antibodies directed against polymerases. There are ~10,000 factories in the nucleoplasm of a HeLa cell, among which are ~8,000 polymerase II factories and ~2,000 polymerase III factories. Each polymerase II factory contains ~8 polymerases. As most active transcription units are associated with only one polymerase, each factory usually contains ~8 different transcription units. These units might be associated through promoters and/or enhancers, with loops forming a "cloud" around the factor.[54]
History
A molecule that allows the genetic material to be realized as a protein was first hypothesized by François Jacob and Jacques Monod. Severo Ochoa won a Nobel Prize in Physiology or Medicine in 1959 for developing a process for synthesizing RNA in vitro with polynucleotide phosphorylase, which was useful for cracking the genetic code. RNA synthesis by RNA polymerase was established in vitro by several laboratories by 1965; however, the RNA synthesized by these enzymes had properties that suggested the existence of an additional factor needed to terminate transcription correctly.
In 1972, Walter Fiers became the first person to actually prove the existence of the terminating enzyme.
Roger D. Kornberg won the 2006 Nobel Prize in Chemistry "for his studies of the molecular basis of eukaryotic transcription".[55]
Measuring and detecting
Transcription can be measured and detected in a variety of ways:
- G-Less Cassette transcription assay: measures promoter strength
- Run-off transcription assay: identifies transcription start sites (TSS)
- Nuclear run-on assay: measures the relative abundance of newly formed transcripts
- KAS-seq: measures single-stranded DNA generated by RNA polymerases; can work with 1,000 cells.[56]
- RNase protection assay and ChIP-Chip of RNAP: detect active transcription sites
- RT-PCR: measures the absolute abundance of total or nuclear RNA levels, which may however differ from transcription rates
- DNA microarrays: measures the relative abundance of the global total or nuclear RNA levels; however, these may differ from transcription rates
- In situ hybridization: detects the presence of a transcript
- MS2 tagging: by incorporating RNA stem loops, such as MS2, into a gene, these become incorporated into newly synthesized RNA. The stem loops can then be detected using a fusion of GFP and the MS2 coat protein, which has a high affinity, sequence-specific interaction with the MS2 stem loops. The recruitment of GFP to the site of transcription is visualized as a single fluorescent spot. This new approach has revealed that transcription occurs in discontinuous bursts, or pulses (see Transcriptional bursting). With the notable exception of in situ techniques, most other methods provide cell population averages, and are not capable of detecting this fundamental property of genes.[57]
- Northern blot: the traditional method, and until the advent of RNA-Seq, the most quantitative
- RNA-Seq: applies next-generation sequencing techniques to sequence whole transcriptomes, which allows the measurement of relative abundance of RNA, as well as the detection of additional variations such as fusion genes, post-transcriptional edits and novel splice sites
- Single cell RNA-Seq: amplifies and reads partial transcriptomes from isolated cells, allowing for detailed analyses of RNA in tissues, embryos, and cancers
Reverse transcription
Some viruses (such as HIV, the cause of AIDS), have the ability to transcribe RNA into DNA. HIV has an RNA genome that is reverse transcribed into DNA. The resulting DNA can be merged with the DNA genome of the host cell. The main enzyme responsible for synthesis of DNA from an RNA template is called reverse transcriptase.
In the case of HIV, reverse transcriptase is responsible for synthesizing a complementary DNA strand (cDNA) to the viral RNA genome. The enzyme ribonuclease H then digests the RNA strand, and reverse transcriptase synthesises a complementary strand of DNA to form a double helix DNA structure ("cDNA"). The cDNA is integrated into the host cell's genome by the enzyme integrase, which causes the host cell to generate viral proteins that reassemble into new viral particles. In HIV, subsequent to this, the host cell undergoes programmed cell death, or apoptosis of T cells.[58] However, in other retroviruses, the host cell remains intact as the virus buds out of the cell.
Some eukaryotic cells contain an enzyme with reverse transcription activity called telomerase. Telomerase is a reverse transcriptase that lengthens the ends of linear chromosomes. Telomerase carries an RNA template from which it synthesizes a repeating sequence of DNA, or "junk" DNA. This repeated sequence of DNA is called a telomere and can be thought of as a "cap" for a chromosome. It is important because every time a linear chromosome is duplicated, it is shortened. With this "junk" DNA or "cap" at the ends of chromosomes, the shortening eliminates some of the non-essential, repeated sequence rather than the protein-encoding DNA sequence, that is farther away from the chromosome end.
Telomerase is often activated in cancer cells to enable cancer cells to duplicate their genomes indefinitely without losing important protein-coding DNA sequence. Activation of telomerase could be part of the process that allows cancer cells to become immortal. The immortalizing factor of cancer via telomere lengthening due to telomerase has been proven to occur in 90% of all carcinogenic tumors in vivo with the remaining 10% using an alternative telomere maintenance route called ALT or Alternative Lengthening of Telomeres.[59]
See also
- Life
- Cell (biology)
- Cell division
- DBTSS
- gene
- gene regulation
- gene expression
- Epigenetics
- Genome
- Crick's central dogma, in which the product of transcription, mRNA, is translated to form polypeptides, and where it is asserted that the reverse processes never occur
- Gene regulation
- Long non-coding RNA
- Missense mRNA
- Splicing - process of removing introns from precursor messenger RNA (pre-mRNA) to make messenger RNA (mRNA)
- Transcriptomics
- Translation (biology)
References
- "RNA Quality and RNA Sample Assessment - US". www.thermofisher.com. Retrieved 2022-08-30.
- Li J, Liu C (2019). "Coding or Noncoding, the Converging Concepts of RNAs". Front Genet. 10: 496. doi:10.3389/fgene.2019.00496. PMC 6538810. PMID 31178900.
- Eldra P. Solomon, Linda R. Berg, Diana W. Martin. Biology, 8th Edition, International Student Edition. Thomson Brooks/Cole. ISBN 978-0495317142
- Koonin EV, Gorbalenya AE, Chumakov KM (July 1989). "Tentative identification of RNA-dependent RNA polymerases of dsRNA viruses and their relationship to positive strand RNA viral polymerases". FEBS Letters. 252 (1–2): 42–6. doi:10.1016/0014-5793(89)80886-5. PMID 2759231. S2CID 36482110.
- "DNA Strands". www.sci.sdsu.edu. Archived from the original on 27 October 2017. Retrieved 1 May 2018.
- Berg J, Tymoczko JL, Stryer L (2006). Biochemistry (6th ed.). San Francisco: W. H. Freeman. ISBN 0-7167-8724-5.
- Watson JD, Baker TA, Bell SP, Gann AA, Levine M, Losick RM (2013). Molecular Biology of the Gene (7th ed.). Pearson.
- Haberle V, Stark A (October 2018). "Eukaryotic core promoters and the functional basis of transcription initiation". Nat Rev Mol Cell Biol. 19 (10): 621–637. doi:10.1038/s41580-018-0028-8. PMC 6205604. PMID 29946135.
- Verheul TC, van Hijfte L, Perenthaler E, Barakat TS (2020). "The Why of YY1: Mechanisms of Transcriptional Regulation by Yin Yang 1". Front Cell Dev Biol. 8: 592164. doi:10.3389/fcell.2020.592164. PMC 7554316. PMID 33102493.
- Spitz F, Furlong EE (September 2012). "Transcription factors: from enhancer binding to developmental control". Nat Rev Genet. 13 (9): 613–26. doi:10.1038/nrg3207. PMID 22868264. S2CID 205485256.
- Beagan JA, Pastuzyn ED, Fernandez LR, Guo MH, Feng K, Titus KR, Chandrashekar H, Shepherd JD, Phillips-Cremins JE (June 2020). "Three-dimensional genome restructuring across timescales of activity-induced neuronal gene expression". Nat Neurosci. 23 (6): 707–717. doi:10.1038/s41593-020-0634-6. PMC 7558717. PMID 32451484.
- Schoenfelder S, Fraser P (August 2019). "Long-range enhancer-promoter contacts in gene expression control". Nat Rev Genet. 20 (8): 437–455. doi:10.1038/s41576-019-0128-0. PMID 31086298. S2CID 152283312.
- Pennacchio LA, Bickmore W, Dean A, Nobrega MA, Bejerano G (April 2013). "Enhancers: five essential questions". Nat Rev Genet. 14 (4): 288–95. doi:10.1038/nrg3458. PMC 4445073. PMID 23503198.
- Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, Guo YE, Hnisz D, Jaenisch R, Bradner JE, Gray NS, Young RA (December 2017). "YY1 Is a Structural Regulator of Enhancer-Promoter Loops". Cell. 171 (7): 1573–1588.e28. doi:10.1016/j.cell.2017.11.008. PMC 5785279. PMID 29224777.
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, Weirauch MT (February 2018). "The Human Transcription Factors". Cell. 172 (4): 650–665. doi:10.1016/j.cell.2018.01.029. PMID 29425488.
- Grossman SR, Engreitz J, Ray JP, Nguyen TH, Hacohen N, Lander ES (July 2018). "Positional specificity of different transcription factor classes within enhancers". Proc Natl Acad Sci U S A. 115 (30): E7222–E7230. doi:10.1073/pnas.1804663115. PMC 6065035. PMID 29987030.
- Allen BL, Taatjes DJ (March 2015). "The Mediator complex: a central integrator of transcription". Nat Rev Mol Cell Biol. 16 (3): 155–66. doi:10.1038/nrm3951. PMC 4963239. PMID 25693131.
- Mikhaylichenko O, Bondarenko V, Harnett D, Schor IE, Males M, Viales RR, Furlong EE (January 2018). "The degree of enhancer or promoter activity is reflected by the levels and directionality of eRNA transcription". Genes Dev. 32 (1): 42–57. doi:10.1101/gad.308619.117. PMC 5828394. PMID 29378788.
- Li QJ, Yang SH, Maeda Y, Sladek FM, Sharrocks AD, Martins-Green M (January 2003). "MAP kinase phosphorylation-dependent activation of Elk-1 leads to activation of the co-activator p300". EMBO J. 22 (2): 281–91. doi:10.1093/emboj/cdg028. PMC 140103. PMID 12514134.
- Carullo NV, Phillips I RA, Simon RC, Soto SA, Hinds JE, Salisbury AJ, Revanna JS, Bunner KD, Ianov L, Sultan FA, Savell KE, Gersbach CA, Day JJ (September 2020). "Enhancer RNAs predict enhancer-gene regulatory links and are critical for enhancer function in neuronal systems". Nucleic Acids Res. 48 (17): 9550–9570. doi:10.1093/nar/gkaa671. PMC 7515708. PMID 32810208.
- Lövkvist C, Dodd IB, Sneppen K, Haerter JO (June 2016). "DNA methylation in human epigenomes depends on local topology of CpG sites". Nucleic Acids Res. 44 (11): 5123–32. doi:10.1093/nar/gkw124. PMC 4914085. PMID 26932361.
- Jabbari K, Bernardi G (May 2004). "Cytosine methylation and CpG, TpG (CpA) and TpA frequencies". Gene. 333: 143–9. doi:10.1016/j.gene.2004.02.043. PMID 15177689.
- Steinhaus R, Gonzalez T, Seelow D, Robinson PN (June 2020). "Pervasive and CpG-dependent promoter-like characteristics of transcribed enhancers". Nucleic Acids Res. 48 (10): 5306–5317. doi:10.1093/nar/gkaa223. PMC 7261191. PMID 32338759.
- Bird A (January 2002). "DNA methylation patterns and epigenetic memory". Genes Dev. 16 (1): 6–21. doi:10.1101/gad.947102. PMID 11782440.
- Du Q, Luu PL, Stirzaker C, Clark SJ (2015). "Methyl-CpG-binding domain proteins: readers of the epigenome". Epigenomics. 7 (6): 1051–73. doi:10.2217/epi.15.39. PMID 25927341.
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM (April 2009). "A census of human transcription factors: function, expression and evolution". Nat. Rev. Genet. 10 (4): 252–63. doi:10.1038/nrg2538. PMID 19274049. S2CID 3207586.
- Sun Z, Xu X, He J, Murray A, Sun MA, Wei X, Wang X, McCoig E, Xie E, Jiang X, Li L, Zhu J, Chen J, Morozov A, Pickrell AM, Theus MH, Xie H (August 2019). "EGR1 recruits TET1 to shape the brain methylome during development and upon neuronal activity". Nat Commun. 10 (1): 3892. Bibcode:2019NatCo..10.3892S. doi:10.1038/s41467-019-11905-3. PMC 6715719. PMID 31467272.
- Kubosaki A, Tomaru Y, Tagami M, Arner E, Miura H, Suzuki T, Suzuki M, Suzuki H, Hayashizaki Y (2009). "Genome-wide investigation of in vivo EGR-1 binding sites in monocytic differentiation". Genome Biol. 10 (4): R41. doi:10.1186/gb-2009-10-4-r41. PMC 2688932. PMID 19374776.
- Bayraktar G, Kreutz MR (April 2018). "Neuronal DNA Methyltransferases: Epigenetic Mediators between Synaptic Activity and Gene Expression?". Neuroscientist. 24 (2): 171–185. doi:10.1177/1073858417707457. PMC 5846851. PMID 28513272.
- Oliveira AM, Hemstedt TJ, Bading H (July 2012). "Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities". Nat Neurosci. 15 (8): 1111–3. doi:10.1038/nn.3151. PMID 22751036. S2CID 10590208.
- Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A (August 2010). "The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation". J Biol Chem. 285 (34): 26114–20. doi:10.1074/jbc.M109.089433. PMC 2924014. PMID 20547484.
- Manzo M, Wirz J, Ambrosi C, Villaseñor R, Roschitzki B, Baubec T (December 2017). "Isoform-specific localization of DNMT3A regulates DNA methylation fidelity at bivalent CpG islands". EMBO J. 36 (23): 3421–3434. doi:10.15252/embj.201797038. PMC 5709737. PMID 29074627.
- Dukatz M, Holzer K, Choudalakis M, Emperle M, Lungu C, Bashtrykov P, Jeltsch A (December 2019). "H3K36me2/3 Binding and DNA Binding of the DNA Methyltransferase DNMT3A PWWP Domain Both Contribute to its Chromatin Interaction". J Mol Biol. 431 (24): 5063–5074. doi:10.1016/j.jmb.2019.09.006. PMID 31634469. S2CID 204832601.
- Bayraktar G, Yuanxiang P, Confettura AD, Gomes GM, Raza SA, Stork O, Tajima S, Suetake I, Karpova A, Yildirim F, Kreutz MR (November 2020). "Synaptic control of DNA methylation involves activity-dependent degradation of DNMT3A1 in the nucleus". Neuropsychopharmacology. 45 (12): 2120–2130. doi:10.1038/s41386-020-0780-2. PMC 7547096. PMID 32726795.
- Boutard, Magali (2016). "Global repositioning of transcription start sites in a plant-fermenting bacterium". Nature Communications. 7: 13783. Bibcode:2016NatCo...713783B. doi:10.1038/ncomms13783. PMC 5171806. PMID 27982035.
- Roeder, Robert G. (1991). "The complexities of eukaryotic transcription initiation: regulation of preinitiation complex assembly". Trends in Biochemical Sciences. 16 (11): 402–408. doi:10.1016/0968-0004(91)90164-Q. ISSN 0968-0004. PMID 1776168.
- Goldman SR, Ebright RH, Nickels BE (May 2009). "Direct detection of abortive RNA transcripts in vivo". Science. 324 (5929): 927–8. Bibcode:2009Sci...324..927G. doi:10.1126/science.1169237. PMC 2718712. PMID 19443781.
- Revyakin A, Liu C, Ebright RH, Strick TR (November 2006). "Abortive initiation and productive initiation by RNA polymerase involve DNA scrunching". Science. 314 (5802): 1139–43. Bibcode:2006Sci...314.1139R. doi:10.1126/science.1131398. PMC 2754787. PMID 17110577.
- Raffaelle M, Kanin EI, Vogt J, Burgess RR, Ansari AZ (November 2005). "Holoenzyme switching and stochastic release of sigma factors from RNA polymerase in vivo". Molecular Cell. 20 (3): 357–66. doi:10.1016/j.molcel.2005.10.011. PMID 16285918.
- Mandal SS, Chu C, Wada T, Handa H, Shatkin AJ, Reinberg D (May 2004). "Functional interactions of RNA-capping enzyme with factors that positively and negatively regulate promoter escape by RNA polymerase II". Proceedings of the National Academy of Sciences of the United States of America. 101 (20): 7572–7. Bibcode:2004PNAS..101.7572M. doi:10.1073/pnas.0401493101. PMC 419647. PMID 15136722.
- Goodrich JA, Tjian R (April 1994). "Transcription factors IIE and IIH and ATP hydrolysis direct promoter clearance by RNA polymerase II". Cell. 77 (1): 145–56. doi:10.1016/0092-8674(94)90242-9. PMID 8156590. S2CID 24602504.
- Milo, Ron; Philips, Rob. "Cell Biology by the Numbers: What is faster, transcription or translation?". book.bionumbers.org. Archived from the original on 20 April 2017. Retrieved 8 March 2017.
- Hodges C, Bintu L, Lubkowska L, Kashlev M, Bustamante C (July 2009). "Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II". Science. 325 (5940): 626–8. Bibcode:2009Sci...325..626H. doi:10.1126/science.1172926. PMC 2775800. PMID 19644123.
- Fitz V, Shin J, Ehrlich C, Farnung L, Cramer P, Zaburdaev V, Grill SW (2016). "Nucleosomal arrangement affects single-molecule transcription dynamics". Proceedings of the National Academy of Sciences. 113 (45): 12733–12738. doi:10.1073/pnas.1602764113. PMC 5111697. PMID 27791062.
- Richardson JP (September 2002). "Rho-dependent termination and ATPases in transcript termination". Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression. 1577 (2): 251–260. doi:10.1016/S0167-4781(02)00456-6. PMID 12213656.
- Lykke-Andersen S, Jensen TH (October 2007). "Overlapping pathways dictate termination of RNA polymerase II transcription". Biochimie. 89 (10): 1177–82. doi:10.1016/j.biochi.2007.05.007. PMID 17629387.
- Cramer, P.; Armache, K.-J.; Baumli, S.; Benkert, S.; Brueckner, F.; Buchen, C.; Damsma, G.E.; Dengl, S.; Geiger, S.R.; Jasiak, A.J.; Jawhari, A. (June 2008). "Structure of Eukaryotic RNA Polymerases". Annual Review of Biophysics. 37 (1): 337–352. doi:10.1146/annurev.biophys.37.032807.130008. ISSN 1936-122X. PMID 18573085.
- http://www.sigmaaldrich.com/US/en/product/sial/h6878 8-Hydroxyquinoline from SIGMA-ALDRICH. Retrieved 2022-02-15
- Datan E, Minn I, Peng X, He QL, Ahn H, Yu B, Pomper MG, Liu JO (2020). "A Glucose-Triptolide Conjugate Selectively Targets Cancer Cells under Hypoxia". iScience. 23 (9): 101536. Bibcode:2020iSci...23j1536D. doi:10.1016/j.isci.2020.101536. PMC 7509213. PMID 33083765.
- Saxonov S, Berg P, Brutlag DL (January 2006). "A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters". Proceedings of the National Academy of Sciences of the United States of America. 103 (5): 1412–7. Bibcode:2006PNAS..103.1412S. doi:10.1073/pnas.0510310103. PMC 1345710. PMID 16432200.
- Bird A (January 2002). "DNA methylation patterns and epigenetic memory". Genes & Development. 16 (1): 6–21. doi:10.1101/gad.947102. PMID 11782440.
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (March 2013). "Cancer genome landscapes". Science. 339 (6127): 1546–58. Bibcode:2013Sci...339.1546V. doi:10.1126/science.1235122. PMC 3749880. PMID 23539594.
- Tessitore A, Cicciarelli G, Del Vecchio F, Gaggiano A, Verzella D, Fischietti M, Vecchiotti D, Capece D, Zazzeroni F, Alesse E (2014). "MicroRNAs in the DNA Damage/Repair Network and Cancer". International Journal of Genomics. 2014: 820248. doi:10.1155/2014/820248. PMC 3926391. PMID 24616890.
- Papantonis A, Kohro T, Baboo S, Larkin JD, Deng B, Short P, Tsutsumi S, Taylor S, Kanki Y, Kobayashi M, Li G, Poh HM, Ruan X, Aburatani H, Ruan Y, Kodama T, Wada Y, Cook PR (November 2012). "TNFα signals through specialized factories where responsive coding and miRNA genes are transcribed". The EMBO Journal. 31 (23): 4404–14. CiteSeerX 10.1.1.919.1919. doi:10.1038/emboj.2012.288. PMC 3512387. PMID 23103767.
- "Chemistry 2006". Nobel Foundation. Archived from the original on March 15, 2007. Retrieved March 29, 2007.
- Wu, T (April 2020). "Kethoxal-assisted single-stranded DNA sequencing captures global transcription dynamics and enhancer activity in situ". Nature Methods. 17 (5): 515–523. doi:10.1038/s41592-020-0797-9. PMC 7205578. PMID 32251394. S2CID 214810294.
- Raj A, van Oudenaarden A (October 2008). "Nature, nurture, or chance: stochastic gene expression and its consequences". Cell. 135 (2): 216–26. doi:10.1016/j.cell.2008.09.050. PMC 3118044. PMID 18957198.
- Kolesnikova IN (2000). "Some patterns of apoptosis mechanism during HIV-infection". Dissertation (in Russian). Archived from the original on July 10, 2011. Retrieved February 20, 2011.
- Cesare AJ, Reddel RR (May 2010). "Alternative lengthening of telomeres: models, mechanisms and implications". Nature Reviews Genetics. 11 (5): 319–30. doi:10.1038/nrg2763. PMID 20351727. S2CID 19224032.
External links
- Interactive Java simulation of transcription initiation. Archived 2011-07-22 at the Wayback Machine From Center for Models of Life at the Niels Bohr Institute.
- Interactive Java simulation of transcription interference--a game of promoter dominance in bacterial virus. Archived 2011-08-26 at the Wayback Machine From Center for Models of Life at the Niels Bohr Institute.
- Virtual Cell Animation Collection, Introducing Transcription