Alcaptonuria
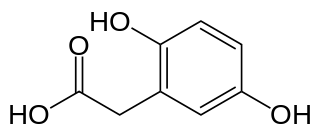
La alcaptonuria (enfermedad de la orina negra u oscura) es una enfermedad hereditaria rara caracterizada por un trastorno del metabolismo de la tirosina y la fenilalanina. Se hereda con un patrón autosómico recesivo y causada por un defecto en el gen HGD que codifica a la enzima homogentisato-1,2-dioxigenasa (EC 1.13.11.5).[1] El resultado es la acumulación de uno de los productos tóxicos de la ruta metabólica en cuestión, una molécula llamada ácido homogentísico, el cual circula por la sangre y se excreta en la orina en grandes cantidades, dándole la característica coloración negruzca a la orina. El exceso del ácido homogentísico causa daño a los cartílagos, una condición llamada ocronosis, que conlleva a osteoartritis, a las válvulas del corazón y produce cálculos renales. La alcaptonuria es frecuente en Eslovaquia y en la República Dominicana, con mayor incidencia que otros países.[2][3]

| Alcaptonuria | ||
|---|---|---|
 | ||
| Especialidad | endocrinología | |
Diagnóstico
El diagnóstico más frecuente se basa en la detección de niveles significativos de ácido homogentísico (HGA) en la orina, mediante análisis cromatográfico. Otra forma de diagnosticar la enfermedad es mediante la secuenciación del gen HGD, para observar si existe alguna mutación en él.
Tratamiento
Es necesario tratar el dolor articular que sufren las personas con alcaptonuria. También es importante una terapia física, para fortalecer la musculatura y la flexibilidad. Si el dolor articular es muy severo, es posible realizar cirugía de reemplazo para aliviarlo. Es recomendable seguir una dieta pobre en fenilalanina y tirosina, pero rica en ácido ascórbico.
Está en estudio el tratamiento por administración de nitisinona, el cual suprime la producción del ácido homogentísico.[4]
Véase también
Referencias
- por MedlinePlus (julio de 2007). «Alcaptonuria». Enciclopedia médica en español. Consultado el 11 de julio de 2008.
- Zatková A, de Bernabé DB, Poláková H, et al (2000). «High frequency of alkaptonuria in Slovakia: evidence for the appearance of multiple mutations in HGO involving different mutational hot spots». American Journal of Human Genetics 67 (5): 1333-9. PMID 11017803.
- Milch RA (1960). «Studies of Alcaptonuria: Inheritance of 47 Cases in Eight Highly Inter-related Dominican Kindreds». Am. J. Hum. Genet. 12 (1): 76-85. PMID 17948450.
- Phornphutkul C, Introne WJ, Perry MB, et al (2002). «Natural history of alkaptonuria». New England Journal Medicine 347 (26): 2111-21. PMID 12501223. doi:10.1056/NEJMoa021736.
Enlaces externos
- Alcaptonuria, Enciclopedia Microsoft® Encarta® Online 2008.
- Entrada en el NCBI sobre la alcaptonuria
- Página en Orphanet
 Wikilibros alberga un libro o manual sobre Alcaptonuria.
Wikilibros alberga un libro o manual sobre Alcaptonuria.
| Control de autoridades |
|
|---|
 Datos: Q651680
Datos: Q651680 Multimedia: Alkaptonuria / Q651680
Multimedia: Alkaptonuria / Q651680