Amiloidosis cardíaca
La amiloidosis cardíaca es una subcategoría de la amiloidosis en la que se produce un depósito de la proteína amiloide en el músculo cardíaco y los tejidos circundantes. El amiloide, una proteína mal plegada e insoluble, puede depositarse en las aurículas, las válvulas o los ventrículos del corazón. Estos depósitos pueden causar el engrosamiento de diferentes secciones del corazón, lo que conduce a una disminución de la función cardíaca[1]. La disminución general de la función cardíaca conduce a una plétora de síntomas.[2] Esta enfermedad multisistémica solía diagnosticarse erróneamente, y anteriormente el diagnóstico se realizaba tras la muerte, durante la autopsia. Sin embargo, los recientes avances tecnológicos han aumentado el diagnóstico de la enfermedad. La amiloidosis cardiaca tiene múltiples subtipos, como la primaria o de cadena ligera (AL), la hereditaria y la senil.[3] Uno de los tipos más estudiados es la amiloidosis cardíaca primaria.[2] El pronóstico depende de la extensión de los depósitos en el organismo y del tipo de amiloidosis.[4] Se están investigando activamente nuevos métodos de tratamiento para la insuficiencia cardíaca y problemas específicos de amiloidosis cardíaca.[5][6]
Tipos
Los diferentes subtipos de amiloidosis cardiaca presentan características epidemiológicas, diagnósticas y pronósticas variables.[4]
Primaria o cadena ligera (AL-CM)
Esta forma relativamente rara de amiloidosis cardiaca se da en un número estimado de seis a diez casos por cada 1.000.000 de personas.[4] Este subtipo suele afectar a varones mayores de 60 años[4] y es rápidamente progresiva. La patogénesis de esta forma se debe a la agregación de cadenas ligeras de inmunoglobulina lambda.[3] Estas cadenas se crean por una expansión anormal de las células plasmáticas.[3] Con el tiempo, estas cadenas ligeras se depositan en el tejido intersticial dentro del miocardio.[4] Las pruebas diagnósticas incluyen electroforesis de suero y orina,[4] pruebas de laboratorio para la determinación de niveles elevados de troponina y BNP, y ECG que muestran voltajes QRS bajos.[2]
Hereditaria (ATTRm-CM)
Este tipo está causado por mutaciones de proteínas implicadas en la formación de amiloide, como la transtiretina (TTR), el fibrinógeno, la apolipoproteína A1 o la apolipoproteína A2. Debido al múltiple número de causas genéticas potenciales, la incidencia de esta forma es variable. La gran mayoría de los casos de amiloidosis cardiaca hereditaria se siguen presentando después de los 60.[4] Una mutación común es la mutación Val122Ile del gen TTR.[2] Se calcula que entre el 3,5 y el 4% de los afroamericanos de Estados Unidos tienen la mutación Val 122lle.[4] Este tipo de amiloidosis se puede identificar mediante pruebas genéticas para la mutación de la proteína.[4] Para realizar el diagnóstico de amiloidosis cardiaca hereditaria se debe obtener una biopsia con evaluación histológica.[7] En esta evaluación histológica se utilizan tinciones especiales para visualizar los depósitos amiloides.[7] Una de estas tinciones es el rojo Congo, que se une específicamente al depósito amiloide y puede caracterizarse mediante diversos métodos de iluminación.[7] Bajo luz polarizada, los depósitos amiloides muestran una birrefringencia verde manzana patognomónica, y bajo luz normal los depósitos aparecen de color rosa salmón claro.[7] Los síntomas de la amiloidosis hereditaria se centran en problemas neuropatológicos y cardíacos.[3] Las manifestaciones cardíacas de la mutación TTR se presentan con más frecuencia en Estados Unidos.[4]
Tipo salvaje (ATTRwt-CM)
Este tipo se considera la mutación de tipo salvaje que da lugar al desarrollo de depósitos de TTR.[2] Suele afectar a varones mayores de 70 años con la manifestación del síndrome del túnel carpiano.[4] Al igual que los demás subtipos de amiloidosis cardíaca, se requiere una biopsia para el diagnóstico.[4] Sin embargo, el diagnóstico formal de amiloidosis cardiaca senil es un diagnóstico de exclusión.[4] La biopsia con evaluación histológica puede descartar los subtipos de cadena ligera y hereditaria, dejando el diagnóstico en senil.[4] Este tipo suele diagnosticarse erróneamente; sin embargo, el mayor uso de la resonancia magnética cardiaca ha aumentado la tasa de diagnóstico.[2] La gravedad de la enfermedad suele ser menor que en las variantes de cadena ligera y hereditaria,[4] debido a que el tiempo que tardan en acumularse los depósitos de amiloide es mayor en la variante senil.[4]
Síntomas y señales
Los síntomas de la amiloidosis cardíaca son una combinación de insuficiencia cardiaca y depósito de amiloide en otros órganos.[2] El depósito de amiloide en el corazón provoca insuficiencia cardiaca diastólica restrictiva que evoluciona a insuficiencia cardiaca sistólica.[8]
Las manifestaciones cardíacas incluyen:
- Disnea por esfuerzo[2]
- Edema periférico y ascitis[2]
- Derrame pericárdico[2]
- Arritmias (secundarias a la alteración del sistema eléctrico normal del corazón)
- Arritmias auriculares (como la fibrilación auricular)[2]
- Bloqueos cardíacos de primer/segundo grado[2]
- Síncope[2]
- Elevación de las venas del cuello y de la presión venosa yugular[9]
- Isquemia miocárdica/Angina (secundaria al depósito de amiloide en las pequeñas arterias del corazón)[2]
- La demanda miocárdica de oxígeno aumenta en pacientes con amiloidosis cardiaca, independientemente de los cambios en la perfusión coronaria.[10]
En los pacientes con amiloidosis de cadena ligera, puede haber depósitos de amiloide en numerosos órganos diferentes.[2] El depósito de amiloide en otros órganos dificulta el diagnóstico de amiloidosis cardíaca, ya que estas manifestaciones extracardíacas enmascaran el diagnóstico.[2] Las manifestaciones extracardíacas incluyen:
- Macroglosia[2]
- Hematoma periorbitario[2]
- Pérdida del tercer y cuarto ruido cardíaco[3]
- Tromboembolismos[2]
- Neuropatía sensorial simétrica (como el túnel carpiano bilateral)[3]
- Hipotensión postural (secundaria a neuropatía autonómica)[2]
- Síndrome nefrótico (secundario al daño de las cadenas ligeras libres en los riñones/deposición de amiloide en los riñones).[2]
Causas
La causa general de la amiloidosis cardiaca es el mal plegamiento de un precursor proteico específico, dependiendo del tipo de amiloidosis. Entre los precursores proteínicos se encuentran las cadenas ligeras derivadas de inmunoglobulinas y las mutaciones de la transtiretina.[3] El mal plegamiento de la proteína hace que tenga láminas beta-plegadas insolubles,[2] creando un amiloide. El amiloide, la agregación o aglomeración de proteínas, es resistente a la degradación por el organismo. Los amiloides son principalmente fibrillas, aunque también contienen un componente P, apolipoproteína, colágeno, fibronectina y laminina.[2] El componente P, una proteína pentamérica, estabiliza las fibrillas del amiloide, lo que reduce su eliminación del organismo.[1] Los depósitos de amiloides pueden producirse en todo el organismo, incluidos el corazón, el hígado, los riñones, el bazo, las glándulas suprarrenales y los huesos. Los depósitos en el espacio extracelular cardíaco pueden endurecer el corazón, lo que provoca una restricción de los ventrículos.[3] Esta restricción del movimiento ventricular provoca una disminución de la capacidad del corazón para bombear con eficacia, lo que da lugar a los diversos síntomas asociados a la amiloidosis cardíaca.[4]
Diagnóstico
Ecocardiografía
La ecocardiografía es un método seguro y no invasivo que puede utilizarse para evaluar la enfermedad estructural y funcional del corazón.[4] La amiloidosis se presenta con engrosamiento ventricular y valvular, agrandamiento biatrial,[4] patrón de llenado restrictivo, con función sistólica normal a levemente reducida[8] y llenado diastólico disminuido.[4] La ecocardiografía puede utilizarse para evaluar el pronóstico de la enfermedad, midiendo las diferentes tensiones dentro del corazón.[4] La amiloidosis cardiaca produce alteraciones específicas de la funcionalidad del corazón. La ecocardiografía puede utilizarse para detectar este patrón específico (preservación relativa del miocardio apical con disminución de la deformación longitudinal en las secciones media y basal), que tiene una sensibilidad del 90-95% y una especificidad del 80-85% para la amiloidosis cardiaca.[4] La ecocardiografía puede utilizarse para ayudar a los médicos con el diagnóstico; sin embargo, sólo puede utilizarse para sugerir la enfermedad, no para confirmarla, a menos que se trate de una amiloidosis en estadio avanzado.[1]
Electrocardiograma (ECG)
Los ECG de pacientes con amiloidosis cardíaca suelen mostrar un bajo voltaje en las derivaciones de las extremidades, con un eje derecho extremo inusual. Suele haber una onda P normal, aunque puede estar ligeramente prolongada. En los pacientes con amiloidosis de cadena ligera, el patrón del complejo QRS está sesgado,[1] con ondas R pobres en las derivaciones torácicas.[2]
Los ECG Holter pueden utilizarse para identificar arritmias asintomáticas.[2]
Puede haber alteraciones en el electrocardiograma, que muestran bajo voltaje y anomalías de la conducción como bloqueo auriculoventricular o disfunción del nódulo sinusal.[8] La fibrilación auricular (FA) se observa hasta en el 70% de los pacientes en el momento del diagnóstico, y los pacientes suelen presentar frecuencias ventriculares controladas causadas por una enfermedad concomitante del sistema de conducción.[11]
Pruebas de laboratorio
Las pruebas de laboratorio incluyen niveles de urea y creatinina, enzimas hepáticas, glucosa, función tiroidea, hemograma completo y pruebas de coagulación. El análisis del suero y la orina para detectar la presencia de inmunoglobulina monoclonal también se realiza mediante inmunofijación para la detección de la banda monoclonal. La presencia de la banda monoclonal sería compatible con la amiloidosis de cadena ligera. En el caso de la amiloidosis de cadena ligera, puede utilizarse el ensayo de cadena ligera libre de inmunoglobulina en suero para el diagnóstico y seguimiento de la amiloidosis.[1] En la amiloidosis de cadena ligera, puede estar presente un nivel bajo de paraproteína.[3]
Biomarcadores cardíacos
Hay dos biomarcadores cardíacos principales que se utilizan en la evaluación de la amiloidosis cardíaca, la troponina y la prohormona N-terminal del péptido natriurético cerebral (NT-proBNP).[12] Como era de esperar, con el daño y la disfunción cardíacos, puede haber una elevación de estos marcadores en pacientes con amiloidosis cardíaca. Estos marcadores se han incorporado a los diversos sistemas de estadificación/puntuación utilizados por los médicos para determinar la gravedad de la enfermedad y el pronóstico.[12]
Biopsias
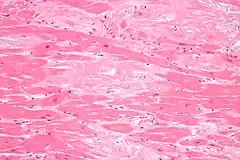
Para confirmar la presencia de depósitos de amiloide pueden utilizarse biopsias extracardíacas de tejidos del riñón, el hígado, el nervio periférico o la grasa abdominal. Los depósitos de amiloide en muestras de biopsia se confirman mediante el uso de colorante rojo Congo, que produce una birrefringencia verde cuando se observa bajo luz polarizada. También puede realizarse una tinción con rojo Sirio o un examen con microscopio electrónico. La determinación del tipo de amiloide puede realizarse mediante técnicas de etiquetado en inmunohistoquímica y tinción de inmunofluorescencia.[1]
En el caso de los pacientes con amiloidosis de cadena ligera, podrían realizarse biopsias de médula ósea para determinar el porcentaje basal de células plasmáticas y descartar un mieloma múltiple.[3]
Cateterismo
El cateterismo cardíaco derecho es la prueba que se utiliza para detectar presiones ventriculares diastólicas elevadas. Esta prueba es más invasiva y se realizaría tras muestras de biopsia endomiocárdica no concluyentes.[1]
Resonancia magnética cardíaca
La resonancia magnética cardiaca (RMC) es capaz de medir el grosor de distintas zonas del corazón. Esto puede utilizarse para la cuantificación de los depósitos en el corazón.[1] La RMC también muestra la caracterización del tejido miocárdico a través de patrones de realce de gadolinio.[2] Sin embargo, ninguna de las técnicas de RMC es capaz de diferenciar definitivamente la ATTR-CM y la AL-CM.[13]
Para la AL-CM, el 68% de ellos presentan hipertrofia ventricular izquierda simétrica y concéntrica. Por otro lado, en el caso de la ATTR-CM, el 79% presenta hipertrofia ventricular izquierda asimétrica y el 18% hipertrofia ventricular izquierda simétrica y concéntrica.[13]
En las imágenes ponderadas en T1, el edema del corazón puede detectarse con una señal T1 elevada. Mientras tanto, el agrandamiento de las células cardiacas reducirá la señal T1. Utilizando la señal T1, el volumen extracelular (VEC) es útil para determinar el grado de depósito de amiloide alrededor de las células cardiacas y detectar la regresión de los depósitos de amiloide tras el tratamiento. El VEC es mayor en la ATTR-CM que en la AL-CM.[13]
En las imágenes ponderadas en T2, la señal T2 aumenta en la miocarditis aguda (inflamación de los músculos del corazón) y en el infarto de miocardio (ataque al corazón). La señal T2 también está aumentada en la AL-CM y la ATTR-CM, pero la señal es mayor en la AL-CM antes de iniciar la quimioterapia.[13]
El realce tardío de gadolinio (RTG) puede determinar la gravedad del depósito de amiloide en el tejido cardiaco. Cuanto mayor es la señal de RTG, más grave es la afectación cardiaca. Puede dividirse en tres fases: ausencia de RTG, RTG subendocárdico y RTG de espesor total (transmural).[13]
Gammagrafía/radionúclidos
La gammagrafía puede utilizarse para medir la extensión y la distribución del amiloide en todo el organismo, incluidos el hígado, el riñón, el bazo y el corazón.[2] Puede administrarse a un paciente por vía intravenosa un componente P del amiloide sérico radiomarcable y el componente P se acumula en el depósito amiloide de forma proporcional al tamaño del depósito. El marcaje del componente P puede visualizarse con una cámara gamma.[1]
En la actualidad, las exploraciones con radionúclidos de tecnecio permiten diagnosticar con fiabilidad la amiloidosis cardiaca, y algunos métodos de exploración tienen una sensibilidad superior al 99% (pero sólo un 91% de especificidad para la amiloidosis).[14] En este método de obtención de imágenes, se inyecta tecnecio radiomarcado en el organismo, donde se une a los depósitos de amiloide cardiaco.[14] Posteriormente, se realiza una gammagrafía para determinar dónde permanece el trazador, lo que pone de manifiesto los depósitos de amiloide en el corazón.[14] Este método permite realizar un diagnóstico definitivo no invasivo de la amiloidosis cardiaca (antes era necesario realizar una biopsia endomiocárdica).[14]
Espectrometría de masas
La espectrometría de masas puede utilizarse para determinar si se trata de una proteína de cadena ligera o de amiloidosis hereditaria mediante la identificación de la subunidad proteica.[9]
Tratamientos
Los tratamientos difieren según el tipo de amiloidosis presente.[1] La mayoría de los tratamientos tienen como objetivo preservar la función cardiaca y tratar los síntomas de insuficiencia cardiaca.
Tratamiento de la amiloidosis de cadenas ligeras (AL-CM): Dado que la causa de este subtipo de amiloidosis cardíaca es la producción excesiva de cadenas ligeras libres, el objetivo principal del tratamiento es la reducción de la concentración de cadenas ligeras.[5] En el caso de la amiloidosis de cadenas ligeras, se pueden utilizar ensayos de CLL y niveles de NT-proBNP para supervisar la progresión de la amiloidosis y cualquier respuesta a los tratamientos.[1] Una de las principales vías para disminuir la producción de este exceso de cadenas ligeras es eliminar las células anómalas que las producen,[5] para lo cual pueden utilizarse agentes quimioterapéuticos como el melfalán o el bortezomib, que eliminan la línea celular anómala que produce las cadenas ligeras libres.[5] Tras la quimioterapia, puede utilizarse un trasplante de médula ósea para restaurar las líneas celulares normales.[5] Existen medicamentos más recientes (ixazomib, carfilzomib, daratumumab, elotuzumab) en investigación para el tratamiento del mieloma múltiple que pueden ayudar a disminuir la producción de cadenas ligeras libres.[5] Nuevos datos sugieren que el trasplante ortotópico de corazón seguido de melfalán y trasplante de células madre produce resultados similares al trasplante de corazón no indicado para amiloidosis cardiaca.[5] Para tratar las complicaciones, pueden prescribirse medicamentos como midodrina para la neuropatía autonómica, amiodarona para pacientes con fibrilación auricular para prevenir arritmias y warfarina utilizada tras un episodio cardioembólico.[1] Deben evitarse los betabloqueantes debido al síntoma habitual de hipotensión.
Tratamiento de la amiloidosis hereditaria (ATTRm-CM): En los últimos años se han producido avances en el tratamiento de la amiloidosis cardiaca hereditaria/transtiretina que incluyen métodos para suprimir la producción de transtiretina, estabilizar las fibrillas amiloides y medicamentos que pueden destruir las fibrillas ya existentes.[6] Para la amiloidosis hereditaria se pueden prescribir inhibidores de la ECA y betabloqueantes si no hay neuropatía autonómica.[1]
- Supresión de la producción de transtiretina: trasplante de hígado y medicamentos que disminuyen la actividad de los genes de la transtiretina (patisiran e inotersen).[6] En pacientes con mutaciones familiares de transtiretina, el trasplante de hígado puede proporcionar al organismo una fuente de transtiretina normal.[15] Al cambiar la fuente de transtiretina del hígado original que contiene la transtiretina mutada a un hígado sano, no habrá más producción de la proteína anormal.[15] Sin embargo, el trasplante de hígado no revierte la enfermedad.[15] El objetivo del trasplante de hígado es prevenir la deposición adicional de amiloide y evitar que se produzcan nuevos síntomas/complicaciones.[15] Estos medicamentos se unen al ARNm de la transtiretina e impiden la producción de la proteína transtiretina, disminuyendo así la cantidad total de transtiretina que puede acumularse en el organismo.
- Estabilización de la transtiretina anormal: Existen medicamentos que pueden estabilizar la transtiretina plegada normalmente, evitando el plegamiento incorrecto y la posterior deposición amiloide.[6] Entre estos medicamentos se encuentran el Tafamidis, el AINE Diflunisal y el AG10.[6] Tafamidis es un medicamento que se une a la transtiretina y la mantiene en su forma normal, impidiendo que se agregue en fibrillas amiloides.[6] Diflunisal y AG10 funcionan de forma similar a Tafamidis en su capacidad de unirse a la transtiretina y estabilizarla.[16]
- Destrucción de las fibrillas amiloides existentes: Existen múltiples medicamentos que muestran propiedades destructoras del amiloide, Doxiciclina, ácido Tauroursodeoxicólico (TUDCA) y anticuerpos monoclonales.[6]
El uso de marcapasos (tanto marcapasos ventricular derecho como marcapasos biventricular) o desfibriladores cardioversores implantables sigue siendo cuestionable en la amiloidosis cardiaca.[17]
En 2012, Craig Lewis, un tejano de 55 años, se presentó en el Texas Heart Institute con un caso grave de amiloidosis. Se le practicó un trasplante experimental de corazón artificial de flujo continuo que le salvó la vida. Lewis murió 5 semanas después de insuficiencia hepática tras entrar en coma debido a la amiloidosis.[18]
Pronóstico
El pronóstico general depende del grado de disfunción cardíaca. Se han observado peores resultados cuando la ecocardiografía muestra grosor de la pared ventricular izquierda, mala función sistólica y disfunción diastólica grave.[1]
Pronóstico para cadena ligera (AL-CM): En el caso de la amiloidosis de cadena ligera, la detección precoz conlleva la mejor posibilidad de terapias que prolonguen el periodo de remisión.[3] La amiloidosis cardiaca de cadena ligera bien tratada tiene una tasa de supervivencia a los 4 años de alrededor del 90%.[5] En los pacientes que se someten a un trasplante de células madre, el tiempo medio de supervivencia aumenta a 10 años.[5] Se han desarrollado sistemas de estadificación para estratificar la gravedad de la enfermedad, incluido el estadio Mayo Biomarker, que utiliza varios biomarcadores como la troponina I, la troponina T, el BNP y el NT-proBNP, y concentraciones de cadenas ligeras libres.[5]
Pronóstico para hereditaria (ATTRm-CM): Debido a la gran cantidad de variables que intervienen en este subtipo, el pronóstico varía según el tipo específico de amiloidosis cardíaca heredirtaria.[5] Las variables incluyen la mutación de la transtiretina mutante frente a la de tipo salvaje y la edad de inicio de los síntomas.[5] En comparación con la amiloidosis de cadena ligera, el subtipo hereditario evoluciona más lentamente y tiene un pronóstico más favorable.[5] Sin embargo, la mutación Val 122lle (la causa más común de amiloidosis cardiaca hereditaria) tiene una tasa de supervivencia a 4 años del 16% con una duración media de 26 meses.[5] Un retraso en el reconocimiento desempeña un factor importante en esta tasa de supervivencia reducida.[5]
Referencias
- Fikrle, Michal; Paleček, Tomáš; Kuchynka, Petr; Němeček, Eduard; Bauerová, Lenka; Straub, Jan; Ryšavá, Romana (1 de febrero de 2013). «Cardiac amyloidosis: A comprehensive review». Cor et Vasa (en inglés) 55 (1): e60-e75. doi:10.1016/j.crvasa.2012.11.018. Consultado el 15 de julio de 2023.
- Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD (2012). «Updates in Cardiac Amyloidosis: A Review». Journal of the American Heart Association (en inglés). PMC 3487372. PMID 23130126. doi:10.1161/JAHA.111.000364.
- Falk RH, Alexander KM, et al. (2016). «AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy». Journal of the American College of Cardiology (en inglés): 1323–1341. PMID 27634125. doi:10.1016/j.jacc.2016.06.053.
- Bhogal S, Ladia V, Sitwala P, Cook E, Bajaj K, Ramu V, et al. (2018). «Cardiac Amyloidosis: An Updated Review With Emphasis on Diagnosis and Future Directions». Current Problems in Cardiology. (en inglés): 10-34. PMID 29173805. doi:10.1016/j.cpcardiol.2017.04.003.
- «Cardiac amyloidosis: An update on pathophysiology, diagnosis, and treatment». Trends in Cardiovascular Medicine (en inglés): 10-21. PMC 5741539. PMID 28739313. doi:10.1016/j.tcm.2017.07.004.
- Manolis, Antonis S.; Manolis, Antonis A.; Manolis, Theodora A.; Melita, Helen (2019-09). «Cardiac amyloidosis: An underdiagnosed/underappreciated disease». European Journal of Internal Medicine 67: 1-13. ISSN 0953-6205. doi:10.1016/j.ejim.2019.07.022. Consultado el 15 de julio de 2023.
- Ruberg FL, Berk JL (2012). «Transthyretin (TTR) Cardiac Amyloidosis». Circulation (en inglés): 1286-1300. PMC 3501197. PMID 22949539. doi:10.1161/CIRCULATIONAHA.111.078915.
- Falk RH, Comenzo RL, et al. (1997). «The Systemic Amyloidoses». New England Journal of Medicine (en inglés). PMID 9302305. doi:10.1056/NEJM199709253371306.
- Gertz MA, Dispenzieri A, Sher T. «Pathophysiology and treatment of cardiac amyloidosis». Nature Reviews Cardiology (en inglés): 91-102. ISSN 1759-5002. PMID 25311231. doi:10.1038/nrcardio.2014.165.
- Clemmensen TS, Soerensen J, Hansson N, Tolbod LP, Harms HJ, Eiskjær H, et al. (2018). «Myocardial Oxygen Consumption and Efficiency in Patients With Cardiac Amyloidosis». Journal of the American Heart Association (en inglés). ISSN 2047-9980. PMC 6404209. PMID 30571379. doi:10.1161/JAHA.118.009974.
- Hartnett, Jack; Jaber, Wael; Maurer, Mathew; Sperry, Brett; Hanna, Mazen; Collier, Patrick; Patel, Divyang R.; Wazni, Oussama M.; Donnellan, Eoin (2021). «Electrophysiological Manifestations of Cardiac Amyloidosis». JACC: CardioOncology (en inglés): 506-515. PMC 8543134. PMID 34729522. doi:10.1016/j.jaccao.2021.07.010.
- Kyriakou P, Mouselimis D, Tsarouchas A, Rigopoulos A, Bakogiannis C, Noutsias M, et al. (2018). «Diagnosis of cardiac amyloidosis: a systematic review on the role of imaging and biomarkers». BMC Cardiovascular Disorders. (en inglés): 221. ISSN 1471-2261. PMC 6278059. PMID 30509186. doi:10.1186/s12872-018-0952-8.
- Martinez-Naharro, Ana; Baksi, A. John; Hawkins, Philip N.; Fontana, Marianna (2020-07). «Diagnostic imaging of cardiac amyloidosis». Nature Reviews Cardiology (en inglés) 17 (7): 413-426. ISSN 1759-5010. doi:10.1038/s41569-020-0334-7. Consultado el 18 de julio de 2023.
- Gillmore, Julian D.; Maurer, Mathew S.; Falk, Rodney H.; Merlini, Giampaolo; Damy, Thibaud; Dispenzieri, Angela; Wechalekar, Ashutosh D.; Berk, John L. et al. (14 de junio de 2016). «Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis». Circulation (en inglés) 133 (24): 2404-2412. ISSN 0009-7322. doi:10.1161/CIRCULATIONAHA.116.021612. Consultado el 18 de julio de 2023.
- Rocha, Ana; Lobato, Luísa (2015-04). «Reply: Liver transplantation in transthyretin amyloidosis: Issues and challenges». Liver Transplantation (en inglés): n/a-n/a. doi:10.1002/lt.24150. Consultado el 18 de julio de 2023.
- Lohrmann G, Pipilas A, Mussinelli R, Gopal DM, Berk JL, Connors LH, et al. (2020). «Stabilization of Cardiac Function With Diflunisal in Transthyretin (ATTR) Cardiac Amyloidosis». Journal of Cardiac Failure: 753-759. PMID 31805416. doi:10.1016/j.cardfail.2019.11.024.
- Cheung, Christopher C.; Roston, Thomas M.; Andrade, Jason G.; Bennett, Matthew T.; Davis, Margot K. (2020-03). «Arrhythmias in Cardiac Amyloidosis: Challenges in Risk Stratification and Treatment». The Canadian Journal of Cardiology 36 (3): 416-423. ISSN 1916-7075. PMID 32145868. doi:10.1016/j.cjca.2019.11.039. Consultado el 18 de julio de 2023.
- Baum, Dan (29 de febrero de 2012). «No Pulse: How Doctors Reinvented The Human Heart». Popular Science (en inglés estadounidense). Consultado el 18 de julio de 2023.