Campo de fuerza (química)
En el contexto de la química y el modelado molecular, un campo de fuerza es un método computacional que es utilizado para estimar las fuerzas entre los átomos dentro de las moléculas y también entre diferentes moléculas. De forma más precisa, el campo de fuerza se refiere a la forma de la función y los parámetros utilizados para calcular la energía potencial de un sistema de átomos o de partículas granuladas mediante mecánica molecular, dinámica molecular o el método de Montecarlo. Los parámetros para una función de energía seleccionada pueden surgir de experimentos físicos o químicos, cálculos de mecánica cuántica o una combinación de ambos. Los campos de fuerza son potenciales interatómicos y utilizan el mismo concepto de campo de fuerza de la física clásica, con la diferencia de que los parámetros del campo de fuerza químico describen un "paisaje de energía", del cual se derivan las fuerzas que actúan sobre cada partícula como un gradiente de energía potencial respecto a las coordenadas de cada partícula.[1]
Los campos de fuerza de todos los átomos proveen parámetros para cada tipo de átomo en un sistema, incluyendo hidrógenos, mientras que los potenciales interatómicos monoatómicos toman al hidrógeno y al carbono de los grupos metilo y metileno como un centro de interacción único.[2] Los potenciales de granulado, que son utilizados en simulaciones de macromoléculas como proteínas, ácidos nucléicos y complejos multicomponente, sacrifican los detalles químicos a favor de una mayor eficiencia computacional.[3]
Forma de la función
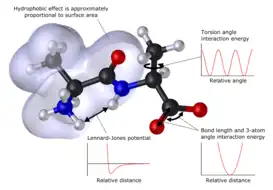
La forma básica de la función de la energía potencial en mecánica molecular incluye términos (llamados enlazantes para interacciones atómicas que están unidas por enlaces covalentes y términos para aquellas interacciones no covalentes (llamados no enlazantes) que describen la interacción electrostática de grupos cargados y la fuerza de van der Waals. La descomposición específica de los términos depende del campo de fuerza, pero una forma general para describir la energía total de un campo de fuerza puede escribirse como la suma de los componentes:

Donde los componentes de cada contribución se desglosan en las siguientes sumas:


El término que describe el enlace y el ángulo son usualmente modelados mediante funciones de energía cuadráticas que no permiten el rompimiento de enlaces. Una descripción más realista de un enlace covalente con un estiramiento alto está dada por el potencial de Morse, aunque requiere de más recursos computacionales para su cálculo. La forma de la función para la energía del diedro es variabla de un campo de fuerza a otro. Adicionalmente, se pueden agregar términos de "torsión impropia" para asegurar la planaridad de anillos aromáticos y otros sistemas conjugados, así como términos "cruzados" que describen el acoplamiento de diferentes variables internas como los ángulos y las longitudes de enlace. Algunos campos de fuerza incluso incluyen términos explícitos para los puentes de hidrógeno.
Los términos no enlazantes son más intensivos computacionalmente. Una elección popular es limitar las interacciones a energías por pares, es decir que, por ejemplo, un catión sólo interactúa con un anión en vez de con todos los aniones presentes. El término de la energía de van der Waals es usualmente calculado con el potencial de Lennard-Jones, mientras que el término electrostático se calcula con la ley de Coulomb, a pesar de que ambos pueden ser amortiguados o amplificados por un factor constante para considerar la polarizabilidad electrónica. Los estudios con estas expresiones de la energía se han enfocado en el estudio de biomoléculas desde la década de 1970 y fueron generalizados a todos los compuestos a inicios del 2000, incluyendo metales, cerámicos, minerales y compuestos orgánicos.[4]
Estiramiento del enlace
A pesar de que es raro que un enlace se desvíe significativamente de sus valores de referencia, la aproximación más simple utiliza la ley de Hooke:

Donde es la constante del resorte, es la longitud del enlace y es la longitud del enlace entre los átomos y cuando todos los otros términos en el campo de fuerza involucrado valen cero. Este último término usualmente es nombrado "longitud de enlace al equilibrio", lo que puede causar confusión. La longitud de enlace al equilibrio es el valor adoptado al equilibrio a 298 K con todos los demás términos del campo de fuerza y energía cinética presentes. Entonces, suele ser apenas diferente del valor de la longitud del enlace en los experimentos a 298 K.[4]





La constante del resorte puede ser determinada a partir de experimentos de espectroscopía infrarroja, espectroscopía Raman o cálculos mecano-cuánticos de alta energía. La constante determina la frecuencia vibracional en simulaciones de dinámica molecular. Mientras más fuerte es el enlace entre los átomos, mayor es el valor de la constante de fuerza y, en consecuencia, mayor es el número de onda en el espectro IR/Raman. [5]
A pesar de que la ley de Hooke provee un nivel aceptable de exactitud, es menos exacto si se aleja de la distancia de equilibrio. Para modelar una curva de Morse, se requiere utilizar potencias cúbicas o superiores.[2][6] Sin embargo, para aplicaciones más prácticas, estas diferencias son despreciables y la exactitud de las predicciones se encuentra en el orden de las milésimas de angstrom, lo cual también es el límite de la confiabilidad de los campos de fuerza comunes. Un potencial de Morse puede ser empleado en su lugar para considerar el rompimiento de los enlaces y ganar una mayor exactitud, a pesar de que es menos eficiente de calcular.
Interacciones electrostáticas
Las interacciones electrostáticas se pueden representar mediante una energía coulómbica, la cual utiliza las cargas atómicas para representar el enlace químico a través del espectro de los enlaces covalentes no polares, covalentes polares e iónicos. La ecuación utilizada es la de la ley de Coulomb:


Donde es la distancia entre los átomos y . La energía total coulómbica es la suma sobre todas las combinaciones pares de átomos y usualmente excluye las interacciones que existen a más de un enlace de distancia.[7][8][9]

Las cargas atómicas pueden hacer contribuciones dominantes a la energía potencial, especialmente en moléculas polares y compuestos iónicos y son críticas para simular la geometría, energía de interacción y la reactividad. La asignación de cargas atómicas usualmente sigue protocolos empíricos y mecano-cuánticos poco fiables, lo que suele conducir a una incertidumbre relativa de hasta el 100% con respecto a valores físicos de momentos dipolares experimentales.[10][11][12] Se han desarrollado cargas atómicas reproducibles para campos de fuerza basados en datos experimentales para deformaciones de densidades electrónicas y momentos dipolares internos.[12][4]
Parametrización
Además de la forma del funcional de los potenciales, los campos de fuerza definen un conjunto de parámetros para diferentes tipos de átomos, enlaces químicos, ángulos diedro, interacciones fuera del plano, interacciones no enlazantes y otros posibles términos.[4] Diversos conjuntos de parámetros son empíricos y algunos campos de fuerza utilizan términos correctivos tan extensos que es difícil asignarles una interpretación física.[13]
Los tipos de átomos se definen para diferentes elementos así como para los mismos elementos en entornos químicos diferentes. Por ejemplo, los átomos de oxígeno en el agua y en el grupo funcional carbonilo son clasificados como diferentes tipos de campos.[14] Los conjuntos de parámetros típicos incluyen valores para la masa atómica, carga iónica, parámetros de Lennard-Jones para cada átomo así como valores al equilibrio de longitudes de enlace, ángulos de enlace y ángulos diedro.[15] Los términos de enlace se refieren a pares, tripletes y cuadrupletes de átomos enlazados e incluyen valores para la constante del resorte efectiva para cada potencial. Los parámetros más recurrentes utilizan un modelo de carga fija que supone que cada átomo posee únicamente un valor asignado para la carga atómica que no se ve afectado por el ambiente electrostático local.[12][16]
La parametrización de os campos para simulaciones de máxima exactitud y transferibilidad siguen un protocolo bien definido[4] que involucra:
- La búsqueda de la estructura cristalina de rayos-x o fórmula química
- La definición de los tipos de átomos
- Definición de las cargas atómicas
- Asignación inicial de los parámetros de enlace y de Lennard-Jones
- Pruebas computacionales de densidad y geometría relativa a los datos experimentales referenciados.
- Pruebas computacionales de propiedades energéticas (energía de superficie,[17] energía de hidratación[18]) relativas a los datos experimentales referenciados.
- Validación secundaria y refinamiento (térmico, mecánico y de propiedades de difusión).[19]
Pueden suceder varios ciclos iterativos durante los pasos 4 y 5, así como entre 6, 4 y 3. La interpretación química de los parámetros y los datos de referencias experimentales juegan un papel crítico.
Los parámetros para simulaciones moleculares de macromoléculas biológicas como proteínas, ADN y ARN usualmente se obtienen de observaciones de moléculas orgánicas pequeñas, que son más accesibles a estudios experimentales y cálculos cuánticos. Por lo tanto, múltiples problemas surgen como:
- Cargas atómicas poco confiables provenientes de cálculos cuánticos que pueden afectar a todas las propiedades calculadas, así como a la consistencia interna.
- Los datos obtenidos de cálculos mecano-cuánticos para moléculas en fase gaseosa pueden no ser transferibles a fases condensadas.
- El uso de datos de moléculas pequeñas en estructuras poliméricas más grandes, involucra incertidumbre en los valores.
- Datos experimentales diferentes con variaciones en la exactitud y los estados de referencia (como la temperatura o fuerza iónica) pueden causar desviaciones. Como resultado, parámetros divergentes para los campos de fuerza se han reportados para moléculas biológicas. Las referencias experimentales incluyen, por ejemplo, la entalpía de vaporización, de sublimación, momentos dipolares y diferentes parámetros espectroscópicos.[20][6][14] Las inconsistencias pueden corregirse mediante la interpretación de todos los parámetros y la elección de un estado de referencia consistente, por ejemplo, presión atmosférica y temperatura estándar.[4]
Diversos campos de fuerza incluyen protocolos poco claros, validaciones incompletas de propiedades clave, falta de interpretación de parámetros y poca discusión de las incertidumbres.[21] En estos casos, amplias y aleatorias desviaciones de las propiedades calculadas se han reportado.
Métodos
Algunos campos de fuerza incluyen modelos explícitos para la polarizabilidad, donde la carga efectiva de una partícula puede ser influenciada por interacciones con sus grupos vecinos. Los modelos de núcleo-coraza son comunes y consisten en una partícula de carga positiva localizada en el núcleo, representando un átomo polarizable y una partícula de carga negativa acoplada al núcleo a través de un potencial de oscilador armónico similar a un resorte.[22][23][24] Ejemplos recientes incluyen modelos polarizables con electrones virtuales que reproducen las cargas en los metales[25] y campos de fuerza de biomoléculas polarizables.[26] Al añadir grados de libertad para la polarizabilidad, la interpretación de los parámetros se vuelve más difícil e incrementa el riesgo de arbitrariedad en la asignación de los parámetros, así como también involucra una disminución en la compatibilidad. Los requerimientos computacionales también se incrementan debido a la necesidad de, repetidamente, calcular el campo electrostático local.
Los modelos polarizables dan buenos resultados cuando capturan cualidades químicas esenciales y la carga atómica neta es relativamente exacta (±10%)[4][27] En tiempos recientes, dichos modelos se han llamado erróneamente "potencial de oscilador de Drude".[28] Un término más apropiado es "modelos de oscilador de Lorentz", ya que fue Lorentz[29] y no Drude[30] quien propuso la forma de acoplar electrones al núcleo.[25] Los modelos de Drude asumen un movimiento sin restricciones de los electrones, como el modelo del gas de electrones para describir metales.[30]
Parametrización
HIstóricamente, diversas aproximaciones a la parametrización se han empleado. Algunos campos de fuerza clásicos recaen en la parametrización poco clara, como el uso de aproximaciones de cálculos mecanocuánticos, usualmente en fase gas, con la esperanza de que exista una correlación con las propiedades en fases condensadas y el uso de modificaciones empíricas que permitan empatar los resultados con datos experimentales.[31][32][33] Sin embargo, este tipo de campos no suelen ser reproducibles, aunado a que su optimización está pensada para ampliar la cobertura y disminuir los requisitos computacionales en vez de optimizarse para tener consistencia química, interpretabilidad y confiabilidad.
De forma similar, algunas herramientas automatizadas se han vuelto disponibles para la parametrización de los campos de fuerza y pueden ayudar a los usuarios a desarrollar sus propios conjuntos de parámetros para campos de la química donde no existen datos.[34][35] Algunos esfuerzos para proveer códigos abiertos incluyen openMM y openMD. El uso de la semi automatización o automatización completa, sin ningún aporte de conocimiento químico, conlleva normalmente un aumento en las inconsistencias a nivel de cargas atómicas. Para la asignación de los parámetros restantes, puede disminuir la interpretabilidad y la función de los parámetros.
Transferibilidad
La forma de los funcionales y los conjuntos de parámetros se han definido por los desarrolladores de potenciales interatómicos e incluye diversos grados de autoconsistencia y transferibilidad. Cuando las formas de los funcionales del potencial varían, los parámetros de un potencial interatómico pueden no ser compatibles con otro funcional.[19] En algunos casos, algunas modificaciones pueden realizarse sin problemas, como por ejemplo, entre los potenciales 9-6-Lennard-Jones para pasar a 12-6-Lennard-Jones.[9] Por el contrario, cuando se intenta pasar de potencial de Buckingham a potencial armónico, se requieren modificaciones adicionales y puede no ser posible la transferencia.
Limitaciones
Todos los potenciales interatómicos están basados en aproximaciones y datos experimentales, por lo que se les nombra "empíricos". El desempeño puede variar desde resultados muy precisos, con cálculos de la Teoría de funcionales de la densidad, hasta simples conjeturas con campos muy simples.[36] El uso de representaciones precisas del enlace químico, combinado con datos experimentales reproducibles y validados, pueden conducir a potenciales interatómicos de alta calidad con muchos menos parámetros y suposiciones comparado con métodos que dependen únicamente de cálculos mecano-cuánticos.[37][38]
Las limitaciones posibles incluyen las cargas atómicas, también llamadas cargas puntuales. La mayoría de los campos de fuerza dependen de cargas puntuales para representar el potencial electrostático alrededor de las moléculas, lo que funciona no tan bien, para distribuciones de carga anisotrópicas.[39] La solución es dar una interpretación clara a las cargas puntuales[12] y adicionar electrones virtuales para capturar las características esenciales de la estructura electrónica, la polarizabilidad adicional en sistemas metálicos, los momentos multipolares internos en sistemas π-conjugados y los pares electrónicos libres en el agua.[40][41][42] La polarización electrónica de los ambientes puede ser descrita al incluir campos de fuerza polarizables,[43][44] o utilizando constantes dieléctricas macroscópicas. Sin embargo, la aplicación de un solo valor de la constante dieléctrica es una aproximación burda a los ambientes altamente heterogéneos de las proteínas, membranas biológicas, minerales o electrolitos.[45]
Fuerzas de Van der Waals
Todos los tipos de fuerzas de Van der Waals también dependen fuertemente del entorno debido a que se originan de interacciones de dipolos instantáneos e inducidos (Interacciones intermoleculares). La teoría original de Fritz London de estas fuerzas es válida únicamente en el vacío. Una teoría más general de las fuerzas de Van der Waals en medios condensados fue propuesta por A.D. McLachlan en 1963 e incluye la aproximación original de London como un caso especial.[46][47] La teoría de McLachlan predice que las atracciones de Van der Waals en medios condensados son más débiles que en el vacío y siguen la regla de "Lo similar disuelve a lo similar", que significa que diferentes tipos de átomos interactúan más débilmente que tipos similares de átomos.[48] Esto se contrasta con las reglas de combinación o la aplicación de la ecuación de Slater-Kirkwood para el desarrollo de campos de fuerza clásicos. Las reglas de combinación establecen que la energía de interacción entre dos átomos diferentes (ej. C-N) es un promedio de las energías de interacción corresopndientes en pares atómicos idénticos (ej. C-C y N-N). De acuerdo con la teoría de McLachlan, la interacción de las partículas en un medio condensado puede ser incluso completamente repulsiva, como se observa con el helio líquido.[46] sin embargo, la falta de vaporización y la presencia de un punto de congelación contradice la teoría de interacciones puramente repulsivas. Las mediciones de las fuerzas de atracción entre diferentes materiales (constante de Hamaker) han sido explicadas por Jacon Israelachvili.[46]
Proteínas
Las limitaciones están más presentes en el refinamiento de la estructura proteica. El mayor reto en este campo es el amplio espacio de conformaciones de las moléculas poliméricas, que requiere de un amplio poder de cómputo cuando la estructura excede los 20 monómeros.[49] Los campos de fuerza han sido aplicados satisfactoriamente en el refinamiento de estructuras proteicas en diferentes aplicaciones para cristalografía de rayos X y espectroscopía de RMN, especialmente con programas como XPLOR.[50] Sin embargo, el refinamiento se lleva a cabo principalmente a través de un conjunto de restricciones experimentales, mientras que los potenciales interatómicos servían principalmente para remover obstáculos interatómicos. Los resultados de los cálculos fueron prácticamente iguales en potenciales esféricos rígidos del programa DYANA[51] (cálculos con datos de RMN) que con programas de refinamiento cristalográfico que no utilizan funciones de energía. Estos problemas están relacionados con los potenciales interatómicos y con la imposibilidad de muestrear el especaio de conformaciones de macromoléculas correctamente.[52] Por lo tanto, el desarrollo de parámetros para describir ese tipo de problemas a gran escala requiere de nuevas aproximaciones. Una área problemática es el modelaje homólogo de proteínas[53] mientras que nuevas funciones empíricas alternativas se han desarrollado para el acoplamiento molecular,[54] plegamiento de proteínas,[55][56][57] refinamiento de modelos,[58] diseño computacional de proteínas,[59][60][61] y el modelaje de proteínas en membranas.[62]
Campos de fuerza ampliamente utilizados
Diferentes campos de fuerza están diseñados para diferentes propósitos. Todos están implementados en diferentes softwares computacionales.
MM2 fue desarrollado por Norman Allinger principalmente para el análisis conformacional de hidrocarburos y otras moléculas orgánicas pequeñas. Está diseñado para reproducir la geometría de equilibrio covalente de moléculas de la manera más precisa posible. Implementa un amplio conjunto de parámetros que es continuamente refinado y actualizado para diferentes tipos de compuestos orgánicos (MM3 y MM4)[63][64][65][66][67]
CFF fue desarrollado por Arieh Warshel, Lifson y sus trabajadores como un método general para unificar estudios de energías, estructuras y vibraciones de moléculas generales y cristales moleculares. El programa CFF, desarrollado por Levitt y Warshel, está basada en una representación cartesiana de todos los átomos y sirvió como base para programas de simulación subsecuentes.
ECEPP fue desarrollado específicamente para el modelaje de péptidos y proteínas. Utiliza geometrías fijas de los residuos de aminoácidos para simplificar las energías potenciales de superficie. Por lo tanto, la minimización de energía se lleva a cabo a través de los ángulos de torsión de la proteína. Tanto MM2 como ECEPP incluyen potenciales para los puentes de hidrógeno y potenciales de torsión para describir rotaciones de enlaces simples. ECEPP/3 fue implementado, con algunas modificaicones, en Internal Coordinate Mechanics (ICM) y FANTOM.[68]
AMBER, CHARMM y GROMOS han sido desarrollados principalmente para la dinámica molecular de macromoléculas, a pesar de que también se utilizan para la minimización de energía. Por lo tanto, las coordenadas de todos los átomos se consideran como variables independientes.
Interface Force Field (IFF)[69] fue desarrollado como el primer campo de fuerza consistente para compuestos de toda la tabla periódica. Sobrepasa las limitaciones conocidas para la asignación de cargas consistentes, utiliza condiciones estándas como el estado de referencia, reproduce estructuras, energías y derivadas de la energía y cuantifica las limitaciones para todos los compuestos incluidos.[4][70] Es compatible con múltiples campos de fuerza para simular materiales híbridos (CHARMM, AMBER, OPLS-AA, CFF, CVFF, GROMOS).
Campos clásicos
- AMBER (Assisted Model Building and Energy Refinement): ampliamente usado para proteínas y ADN.
- CFF (Consistent Force Field): es una familia de campos de fuerza adaptados a una amplia variedad de compuestos orgánicos, incluyendo polímeros y metales.
- CHARMM (Chemistry at Harvard Molecular Mechanics): Originalmente desarrollado en Harvard, ampliamente usado tanto para moléculas pequeñas como macromoléculas
- COSMOS-NMR: campo de fuerza híbrido QM/MM adaptado a compuestos inorgánicos, orgánicos y macromoléculas biológicas, incluyendo cálculos semi-empíricos de cargas atómicas y propiedades de RMN. Está optimizado para la elucidación de estructuras basadas en su espectro de RMN e implementa el paquete de modelaje molecular COSMOS.[71]
- CVFF: también se utiliza ampliamente para moléculas pequeñas y macromoléculas.
- ECEPP: Primer campo de fuerza para polipéptidos, desarrollado por F.A. Momany, H.A. Scheraga et. al. [72][73]
- GROMOS (Groningen Molecular Simulation): es un campo de fuerza parte del software GROMOS, un paquete de simulación de dinámica molecular para propósitos generales y para el estudio sistemas biomoleculares.
- IFF (Interface Force Field)
- MMFF (Merck Molecular Force Field)
- OPLS (Optimized Potential for Liquid Simulations), sus variantes incluyen OPLS-AA, OPLS-UA, OPLS-2001, OPLS-2005, OPLS3e, OPLS4; desarrolladas en el departamento de Química de la Universidad de Yale
- QCFF/PI
- UFF (Universal Force Field)
Polarizables
- AMBER
- AMOEBA (Atomic Multipole Optimized Energetics for Biomolecular Applicationes)
- CHARMM
- CFF/ind y ENZYMIX
- COSMOS-NMR (Computer Simulation of Molecular Structure)
- DRF90
- IFF Incluye algunos campos polarizables para metales y moléculas pi-conjugadas
- NEMO (Non-Empirical Molecular Orbital)
- PIPF
- PFF (Polarizable Force Field)
- CPE (Chemical Potential Equalization)
- PHAST
- ORIENT
- GEM (Gaussian Electrostatic Model)
- APPLE&P (Atomistic Polarizable Potential for Liquids, Electrolytes and Polymers)
- GFN-FF (Geometry, Frequency and Noncovalent Interaction Force Fields)
Reactivos
- EVB (Empirical Valence Bond): es un campo de fuerza reactivo que sirve para modela reacciones químicas en diferentes entornos. Facilita el cálculo de energías de activación en fases condensadas y enzimas.
- ReaxFF (Reactive Force Field)
De grano grueso
- DPD (Dissipative Particle Dynamics)
- MARTINI
- SIRAH
- VAMM
Machine Learning
- ANI
- FFLUX
- TensorMol
- Δ-ML
- SchNet
- PhysNEt
Referencias
- Frenkel, Daan, 1948-. Understanding molecular simulation : from algorithms to applications (2nd ed edición). ISBN 978-0-08-051998-2. OCLC 173686073. Consultado el 1 de diciembre de 2020.
- Leach, Andrew R. (2001). Molecular modelling : principles and applications (2nd ed edición). Prentice Hall. ISBN 0-582-38210-6. OCLC 45008511. Consultado el 1 de diciembre de 2020.
- Marrink, Siewert J.; Risselada, H. Jelger; Yefimov, Serge; Tieleman, D. Peter; de Vries, Alex H. (1 de julio de 2007). «The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations». The Journal of Physical Chemistry B 111 (27): 7812-7824. ISSN 1520-6106. doi:10.1021/jp071097f. Consultado el 1 de diciembre de 2020.
- Heinz, Hendrik; Lin, Tzu-Jen; Kishore Mishra, Ratan; Emami, Fateme S. (12 de febrero de 2013). «Thermodynamically Consistent Force Fields for the Assembly of Inorganic, Organic, and Biological Nanostructures: The INTERFACE Force Field». Langmuir 29 (6): 1754-1765. ISSN 0743-7463. doi:10.1021/la3038846. Consultado el 1 de diciembre de 2020.
- Heinz, Hendrik; Koerner, Hilmar; Anderson, Kelly L.; Vaia, Richard A.; Farmer, B. L. (1 de noviembre de 2005). «Force Field for Mica-Type Silicates and Dynamics of Octadecylammonium Chains Grafted to Montmorillonite». Chemistry of Materials 17 (23): 5658-5669. ISSN 0897-4756. doi:10.1021/cm0509328. Consultado el 17 de diciembre de 2020.
- Sun, Huai; Mumby, Stephen J.; Maple, Jon R.; Hagler, Arnold T. (1 de abril de 1994). «An ab Initio CFF93 All-Atom Force Field for Polycarbonates». Journal of the American Chemical Society 116 (7): 2978-2987. ISSN 0002-7863. doi:10.1021/ja00086a030. Consultado el 17 de diciembre de 2020.
- Huang, Jing; MacKerell, Alexander D. (2013). «CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data». Journal of Computational Chemistry (en inglés) 34 (25): 2135-2145. ISSN 1096-987X. PMC 3800559. PMID 23832629. doi:10.1002/jcc.23354. Consultado el 17 de diciembre de 2020.
- Wang, Junmei; Wolf, Romain M.; Caldwell, James W.; Kollman, Peter A.; Case, David A. (15 de julio de 2004). «Development and testing of a general amber force field». Journal of Computational Chemistry 25 (9): 1157-1174. ISSN 0192-8651. PMID 15116359. doi:10.1002/jcc.20035. Consultado el 17 de diciembre de 2020.
- Mishra, Ratan K.; Fernández-Carrasco, Lucia; Flatt, Robert J.; Heinz, Hendrik (17 de junio de 2014). «A force field for tricalcium aluminate to characterize surface properties, initial hydration, and organically modified interfaces in atomic resolution». Dalton Transactions (en inglés) 43 (27): 10602-10616. ISSN 1477-9234. doi:10.1039/C4DT00438H. Consultado el 17 de diciembre de 2020.
- Gross, Kevin C.; Seybold, Paul G.; Hadad, Christopher M. (2002). «Comparison of different atomic charge schemes for predicting pKa variations in substituted anilines and phenols*». International Journal of Quantum Chemistry (en alemán) 90 (1): 445-458. ISSN 1097-461X. doi:10.1002/qua.10108. Consultado el 17 de diciembre de 2020.
- Wang, Bo; Li, Shaohong L.; Truhlar, Donald G. (9 de diciembre de 2014). «Modeling the Partial Atomic Charges in Inorganometallic Molecules and Solids and Charge Redistribution in Lithium-Ion Cathodes». Journal of Chemical Theory and Computation 10 (12): 5640-5650. ISSN 1549-9618. doi:10.1021/ct500790p. Consultado el 17 de diciembre de 2020.
- Heinz, Hendrik; Suter, Ulrich W. (1 de noviembre de 2004). «Atomic Charges for Classical Simulations of Polar Systems». The Journal of Physical Chemistry B 108 (47): 18341-18352. ISSN 1520-6106. doi:10.1021/jp048142t. Consultado el 17 de diciembre de 2020.
- van Duin, Adri C. T.; Dasgupta, Siddharth; Lorant, Francois; Goddard, William A. (1 de octubre de 2001). «ReaxFF: A Reactive Force Field for Hydrocarbons». The Journal of Physical Chemistry A 105 (41): 9396-9409. ISSN 1089-5639. doi:10.1021/jp004368u. Consultado el 25 de mayo de 2021.
- Dauber-Osguthorpe, Pnina; Roberts, Victoria A.; Osguthorpe, David J.; Wolff, Jon; Genest, Moniqe; Hagler, Arnold T. (1988). «Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductase-trimethoprim, a drug-receptor system». Proteins: Structure, Function, and Bioinformatics (en inglés) 4 (1): 31-47. ISSN 1097-0134. doi:10.1002/prot.340040106. Consultado el 25 de mayo de 2021.
- Dharmawardhana, Chamila C.; Kanhaiya, Krishan; Lin, Tzu-Jen; Garley, Amanda; Knecht, Marc R.; Zhou, Jihan; Miao, Jianwei; Heinz, Hendrik (2 de noviembre de 2017). «Reliable computational design of biological-inorganic materials to the large nanometer scale using Interface-FF». Molecular Simulation 43 (13-16): 1394-1405. ISSN 0892-7022. doi:10.1080/08927022.2017.1332414. Consultado el 25 de mayo de 2021.
- Liu, Juan; Tennessen, Emrys; Miao, Jianwei; Huang, Yu; Rondinelli, James M.; Heinz, Hendrik (5 de julio de 2018). «Understanding Chemical Bonding in Alloys and the Representation in Atomistic Simulations». The Journal of Physical Chemistry C 122 (26): 14996-15009. ISSN 1932-7447. doi:10.1021/acs.jpcc.8b01891. Consultado el 25 de mayo de 2021.
- Heinz, Hendrik; Vaia, R. A.; Farmer, B. L.; Naik, R. R. (6 de noviembre de 2008). «Accurate Simulation of Surfaces and Interfaces of Face-Centered Cubic Metals Using 12−6 and 9−6 Lennard-Jones Potentials». The Journal of Physical Chemistry C 112 (44): 17281-17290. ISSN 1932-7447. doi:10.1021/jp801931d. Consultado el 25 de mayo de 2021.
- Emami, Fateme S.; Puddu, Valeria; Berry, Rajiv J.; Varshney, Vikas; Patwardhan, Siddharth V.; Perry, Carole C.; Heinz, Hendrik (22 de abril de 2014). «Force Field and a Surface Model Database for Silica to Simulate Interfacial Properties in Atomic Resolution». Chemistry of Materials 26 (8): 2647-2658. ISSN 0897-4756. doi:10.1021/cm500365c. Consultado el 25 de mayo de 2021.
- Heinz, Hendrik; Ramezani-Dakhel, Hadi (18 de enero de 2016). «Simulations of inorganic–bioorganic interfaces to discover new materials: insights, comparisons to experiment, challenges, and opportunities». Chemical Society Reviews (en inglés) 45 (2): 412-448. ISSN 1460-4744. doi:10.1039/C5CS00890E. Consultado el 25 de mayo de 2021.
- Jorgensen, William L.; Maxwell, David S.; Tirado-Rives, Julian (13 de noviembre de 1996). «Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids». Journal of the American Chemical Society 118 (45): 11225-11236. ISSN 0002-7863. doi:10.1021/ja9621760. Consultado el 25 de mayo de 2021.
- Rappe, A. K.; Casewit, C. J.; Colwell, K. S.; Goddard, W. A.; Skiff, W. M. (1 de diciembre de 1992). «UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations». Journal of the American Chemical Society 114 (25): 10024-10035. ISSN 0002-7863. doi:10.1021/ja00051a040. Consultado el 25 de mayo de 2021.
- Dick, B. G.; Overhauser, A. W. (1 de octubre de 1958). «Theory of the Dielectric Constants of Alkali Halide Crystals». Physical Review 112 (1): 90-103. doi:10.1103/PhysRev.112.90. Consultado el 25 de mayo de 2021.
- Mitchell, P. J.; Fincham, D. (1993-02). «Shell model simulations by adiabatic dynamics». Journal of Physics: Condensed Matter (en inglés) 5 (8): 1031-1038. ISSN 0953-8984. doi:10.1088/0953-8984/5/8/006. Consultado el 25 de mayo de 2021.
- Yu, Haibo; van Gunsteren, Wilfred F. (1 de noviembre de 2005). «Accounting for polarization in molecular simulation». Computer Physics Communications (en inglés) 172 (2): 69-85. ISSN 0010-4655. doi:10.1016/j.cpc.2005.01.022. Consultado el 25 de mayo de 2021.
- Geada, Isidro Lorenzo; Ramezani-Dakhel, Hadi; Jamil, Tariq; Sulpizi, Marialore; Heinz, Hendrik (19 de febrero de 2018). «Insight into induced charges at metal surfaces and biointerfaces using a polarizable Lennard–Jones potential». Nature Communications (en inglés) 9 (1): 716. ISSN 2041-1723. PMC 5818522. PMID 29459638. doi:10.1038/s41467-018-03137-8. Consultado el 25 de mayo de 2021.
- Patel, Sandeep; Brooks, Charles L. (2004). «CHARMM fluctuating charge force field for proteins: I parameterization and application to bulk organic liquid simulations». Journal of Computational Chemistry (en inglés) 25 (1): 1-16. ISSN 1096-987X. doi:10.1002/jcc.10355. Consultado el 25 de mayo de 2021.
- Lin, Tzu-Jen; Heinz, Hendrik (10 de marzo de 2016). «Accurate Force Field Parameters and pH Resolved Surface Models for Hydroxyapatite to Understand Structure, Mechanics, Hydration, and Biological Interfaces». The Journal of Physical Chemistry C 120 (9): 4975-4992. ISSN 1932-7447. doi:10.1021/acs.jpcc.5b12504. Consultado el 25 de mayo de 2021.
- Lemkul, Justin A.; Huang, Jing; Roux, Benoît; MacKerell, Alexander D. (11 de mayo de 2016). «An Empirical Polarizable Force Field Based on the Classical Drude Oscillator Model: Development History and Recent Applications». Chemical Reviews 116 (9): 4983-5013. ISSN 0009-2665. PMC 4865892. PMID 26815602. doi:10.1021/acs.chemrev.5b00505. Consultado el 25 de mayo de 2021.
- Lorentz, H. A. (1904). «The motion of electrons in metallic bodies I». Koninklijke Nederlandse Akademie van Wetenschappen Proceedings Series B Physical Sciences 7: 438-453. Consultado el 25 de mayo de 2021.
- Drude, P. (1900). «Zur Elektronentheorie der Metalle». Annalen der Physik (en inglés) 306 (3): 566-613. ISSN 1521-3889. doi:10.1002/andp.19003060312. Consultado el 25 de mayo de 2021.
- Siu, Shirley W. I.; Pluhackova, Kristyna; Böckmann, Rainer A. (10 de abril de 2012). «Optimization of the OPLS-AA Force Field for Long Hydrocarbons». Journal of Chemical Theory and Computation 8 (4): 1459-1470. ISSN 1549-9618. doi:10.1021/ct200908r. Consultado el 26 de mayo de 2021.
- Aduri, Raviprasad; Psciuk, Brian T.; Saro, Pirro; Taniga, Hariprakash; Schlegel, H. Bernhard; SantaLucia, John (1 de julio de 2007). «AMBER Force Field Parameters for the Naturally Occurring Modified Nucleosides in RNA». Journal of Chemical Theory and Computation 3 (4): 1464-1475. ISSN 1549-9618. doi:10.1021/ct600329w. Consultado el 26 de mayo de 2021.
- Kirschner, Karl N.; Lins, Roberto D.; Maass, Astrid; Soares, Thereza A. (13 de noviembre de 2012). «A Glycam-Based Force Field for Simulations of Lipopolysaccharide Membranes: Parametrization and Validation». Journal of Chemical Theory and Computation 8 (11): 4719-4731. ISSN 1549-9618. doi:10.1021/ct300534j. Consultado el 26 de mayo de 2021.
- Wang, Lee-Ping; Martinez, Todd J.; Pande, Vijay S. (5 de junio de 2014). «Building Force Fields: An Automatic, Systematic, and Reproducible Approach». The Journal of Physical Chemistry Letters 5 (11): 1885-1891. doi:10.1021/jz500737m. Consultado el 26 de mayo de 2021.
- McDonagh, James L.; Shkurti, Ardita; Bray, David J.; Anderson, Richard L.; Pyzer-Knapp, Edward O. (28 de octubre de 2019). «Utilizing Machine Learning for Efficient Parameterization of Coarse Grained Molecular Force Fields». Journal of Chemical Information and Modeling 59 (10): 4278-4288. ISSN 1549-9596. doi:10.1021/acs.jcim.9b00646. Consultado el 26 de mayo de 2021.
- Emami, Fateme S.; Puddu, Valeria; Berry, Rajiv J.; Varshney, Vikas; Patwardhan, Siddharth V.; Perry, Carole C.; Heinz, Hendrik (22 de abril de 2014). «Force Field and a Surface Model Database for Silica to Simulate Interfacial Properties in Atomic Resolution». Chemistry of Materials 26 (8): 2647-2658. ISSN 0897-4756. doi:10.1021/cm500365c. Consultado el 26 de mayo de 2021.
- Ruiz, Victor G.; Liu, Wei; Tkatchenko, Alexandre (15 de enero de 2016). «Density-functional theory with screened van der Waals interactions applied to atomic and molecular adsorbates on close-packed and non-close-packed surfaces». Physical Review B 93 (3): 035118. doi:10.1103/PhysRevB.93.035118. Consultado el 26 de mayo de 2021.
- Ruiz, Victor G.; Liu, Wei; Zojer, Egbert; Scheffler, Matthias; Tkatchenko, Alexandre (6 de abril de 2012). «Density-Functional Theory with Screened van der Waals Interactions for the Modeling of Hybrid Inorganic-Organic Systems». Physical Review Letters 108 (14): 146103. doi:10.1103/PhysRevLett.108.146103. Consultado el 26 de mayo de 2021.
- Kramer, Christian; Spinn, Alexander; Liedl, Klaus R. (14 de octubre de 2014). «Charge Anisotropy: Where Atomic Multipoles Matter Most». Journal of Chemical Theory and Computation 10 (10): 4488-4496. ISSN 1549-9618. doi:10.1021/ct5005565. Consultado el 28 de mayo de 2021.
- Mahoney, Michael W.; Jorgensen, William L. (5 de mayo de 2000). «A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions». The Journal of Chemical Physics 112 (20): 8910-8922. ISSN 0021-9606. doi:10.1063/1.481505. Consultado el 28 de mayo de 2021.
- Xu, Rui; Chen, Chien-Chun; Wu, Li; Scott, M. C.; Theis, W.; Ophus, Colin; Bartels, Matthias; Yang, Yongsoo et al. (2015-11). «Three-dimensional coordinates of individual atoms in materials revealed by electron tomography». Nature Materials (en inglés) 14 (11): 1099-1103. ISSN 1476-4660. doi:10.1038/nmat4426. Consultado el 28 de mayo de 2021.
- Pramanik, Chandrani; Gissinger, Jacob R.; Kumar, Satish; Heinz, Hendrik (26 de diciembre de 2017). «Carbon Nanotube Dispersion in Solvents and Polymer Solutions: Mechanisms, Assembly, and Preferences». ACS Nano 11 (12): 12805-12816. ISSN 1936-0851. doi:10.1021/acsnano.7b07684. Consultado el 28 de mayo de 2021.
- Lomize, Andrei L.; Reibarkh, Mikhail Y.; Pogozheva, Irina D. (2002). «Interatomic potentials and solvation parameters from protein engineering data for buried residues». Protein Science (en inglés) 11 (8): 1984-2000. ISSN 1469-896X. PMC 2373680. PMID 12142453. doi:10.1110/ps.0307002. Consultado el 28 de mayo de 2021.
- «Modeling electrostatic effects in proteins». Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics (en inglés) 1764 (11): 1647-1676. 1 de noviembre de 2006. ISSN 1570-9639. doi:10.1016/j.bbapap.2006.08.007. Consultado el 28 de mayo de 2021.
- Schutz, Claudia N.; Warshel, Arieh (2001). «What are the dielectric “constants” of proteins and how to validate electrostatic models?». Proteins: Structure, Function, and Bioinformatics (en inglés) 44 (4): 400-417. ISSN 1097-0134. doi:10.1002/prot.1106. Consultado el 28 de mayo de 2021.
- Intermolecular and Surface Forces (en inglés). Academic Press. 1 de enero de 2011. ISBN 978-0-12-391927-4. doi:10.1016/b978-0-12-391927-4.10024-6. Consultado el 28 de mayo de 2021.
- McLachlan, A.D. (1964-01). «Van der Waals forces between an atom and a surface». Molecular Physics (en inglés) 7 (4): 381-388. ISSN 0026-8976. doi:10.1080/00268976300101141. Consultado el 28 de mayo de 2021.
- Leckband, Deborah; Israelachvili, Jacob (2001/05). «Intermolecular forces in biology». Quarterly Reviews of Biophysics (en inglés) 34 (2): 105-267. ISSN 1469-8994. doi:10.1017/S0033583501003687. Consultado el 28 de mayo de 2021.
- Pramanik, Chandrani; Jamil, Tariq; Gissinger, Jacob R.; Guittet, Darice; Arias-Monje, Pedro J.; Kumar, Satish; Heinz, Hendrik (2019). «Polyacrylonitrile Interactions with Carbon Nanotubes in Solution: Conformations and Binding as a Function of Solvent, Temperature, and Concentration». Advanced Functional Materials (en inglés) 29 (50): 1905247. ISSN 1616-3028. doi:10.1002/adfm.201905247. Consultado el 28 de mayo de 2021.
- Brunger, Axel T.; Adams, Paul D. (1 de junio de 2002). «Molecular Dynamics Applied to X-ray Structure Refinement». Accounts of Chemical Research 35 (6): 404-412. ISSN 0001-4842. doi:10.1021/ar010034r. Consultado el 28 de mayo de 2021.
- Güntert, Peter (1998/05). «Structure calculation of biological macromolecules from NMR data». Quarterly Reviews of Biophysics (en inglés) 31 (2): 145-237. ISSN 1469-8994. doi:10.1017/S0033583598003436. Consultado el 28 de mayo de 2021.
- Ostermeir, Katja; Zacharias, Martin (1 de enero de 2013). «163 Enhanced sampling of peptides and proteins with a new biasing replica exchange method». Journal of Biomolecular Structure and Dynamics 31 (sup1): 106-106. ISSN 0739-1102. doi:10.1080/07391102.2013.786405. Consultado el 28 de mayo de 2021.
- Tramontano, Anna; Morea, Veronica (2003). «Assessment of homology-based predictions in CASP5». Proteins: Structure, Function, and Bioinformatics (en inglés) 53 (S6): 352-368. ISSN 1097-0134. doi:10.1002/prot.10543. Consultado el 28 de mayo de 2021.
- Gohlke, Holger; Klebe, Gerhard (2002). «Approaches to the Description and Prediction of the Binding Affinity of Small-Molecule Ligands to Macromolecular Receptors». Angewandte Chemie International Edition (en inglés) 41 (15): 2644-2676. ISSN 1521-3773. doi:10.1002/1521-3773(20020802)41:153.0.CO;2-O. Consultado el 28 de mayo de 2021.
- «Structural energetics of protein folding and binding». Current Opinion in Biotechnology (en inglés) 11 (1): 62-66. 1 de febrero de 2000. ISSN 0958-1669. doi:10.1016/S0958-1669(99)00055-5. Consultado el 28 de mayo de 2021.
- «Effective energy functions for protein structure prediction». Current Opinion in Structural Biology (en inglés) 10 (2): 139-145. 1 de abril de 2000. ISSN 0959-440X. doi:10.1016/S0959-440X(00)00063-4. Consultado el 28 de mayo de 2021.
- Javidpour, Leili (2012-03). «Computer Simulations of Protein Folding». Computing in Science Engineering 14 (2): 97-103. ISSN 1558-366X. doi:10.1109/MCSE.2012.21. Consultado el 28 de mayo de 2021.
- Krieger, Elmar; Joo, Keehyoung; Lee, Jinwoo; Lee, Jooyoung; Raman, Srivatsan; Thompson, James; Tyka, Mike; Baker, David et al. (2009). «Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8». Proteins: Structure, Function, and Bioinformatics (en inglés) 77 (S9): 114-122. ISSN 1097-0134. PMC 2922016. PMID 19768677. doi:10.1002/prot.22570. Consultado el 28 de mayo de 2021.
- «Energy functions for protein design». Current Opinion in Structural Biology (en inglés) 9 (4): 509-513. 1 de agosto de 1999. ISSN 0959-440X. doi:10.1016/S0959-440X(99)80072-4. Consultado el 28 de mayo de 2021.
- «Energy estimation in protein design». Current Opinion in Structural Biology (en inglés) 12 (4): 441-446. 1 de agosto de 2002. ISSN 0959-440X. doi:10.1016/S0959-440X(02)00345-7. Consultado el 28 de mayo de 2021.
- «Protein Structure Prediction Using Rosetta». Methods in Enzymology (en inglés) 383: 66-93. 1 de enero de 2004. ISSN 0076-6879. doi:10.1016/S0076-6879(04)83004-0. Consultado el 28 de mayo de 2021.
- Lomize, Andrei L.; Pogozheva, Irina D.; Lomize, Mikhail A.; Mosberg, Henry I. (2006). «Positioning of proteins in membranes: A computational approach». Protein Science (en inglés) 15 (6): 1318-1333. ISSN 1469-896X. PMC 2242528. PMID 16731967. doi:10.1110/ps.062126106. Consultado el 28 de mayo de 2021.
- Allinger, Norman L. (1 de diciembre de 1977). «Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms». Journal of the American Chemical Society 99 (25): 8127-8134. ISSN 0002-7863. doi:10.1021/ja00467a001. Consultado el 28 de mayo de 2021.
- «Information on MM2/MM3». web.archive.org. 23 de enero de 2009. Archivado desde el original el 23 de enero de 2009. Consultado el 28 de mayo de 2021.
- Allinger, Norman L.; Yuh, Young H.; Lii, Jenn Huei (1 de noviembre de 1989). «Molecular mechanics. The MM3 force field for hydrocarbons. 1». Journal of the American Chemical Society 111 (23): 8551-8566. ISSN 0002-7863. doi:10.1021/ja00205a001. Consultado el 28 de mayo de 2021.
- Lii, Jenn Huei; Allinger, Norman L. (1 de noviembre de 1989). «Molecular mechanics. The MM3 force field for hydrocarbons. 2. Vibrational frequencies and thermodynamics». Journal of the American Chemical Society 111 (23): 8566-8575. ISSN 0002-7863. doi:10.1021/ja00205a002. Consultado el 28 de mayo de 2021.
- Lii, Jenn Huei; Allinger, Norman L. (1 de noviembre de 1989). «Molecular mechanics. The MM3 force field for hydrocarbons. 3. The van der Waals' potentials and crystal data for aliphatic and aromatic hydrocarbons». Journal of the American Chemical Society 111 (23): 8576-8582. ISSN 0002-7863. doi:10.1021/ja00205a003. Consultado el 28 de mayo de 2021.
- Schaumann, Thomas; Braun, Werner; Wüthrich, Kurt (1990). «The program FANTOM for energy refinement of polypeptides and proteins using a Newton – Raphson minimizer in torsion angle space». Biopolymers (en inglés) 29 (4-5): 679-694. ISSN 1097-0282. doi:10.1002/bip.360290403. Consultado el 28 de mayo de 2021.
- «INTERFACE MD». INTERFACES LABORATORY (en inglés). 2 de febrero de 2016. Consultado el 28 de mayo de 2021.
- «cemff: A force field database for cementitious materials including validations, applications and opportunities». Cement and Concrete Research (en inglés) 102: 68-89. 1 de diciembre de 2017. ISSN 0008-8846. doi:10.1016/j.cemconres.2017.09.003. Consultado el 28 de mayo de 2021.
- Möllhoff, Margit; Sternberg, Ulrich (1 de mayo de 2001). «Molecular mechanics with fluctuating atomic charges – a new force field with a semi-empirical charge calculation». Molecular modeling annual (en inglés) 7 (4): 90-102. ISSN 0948-5023. doi:10.1007/s008940100008. Consultado el 28 de mayo de 2021.
- Momany, F. A.; McGuire, R. F.; Burgess, A. W.; Scheraga, Harold A. (1 de octubre de 1975). «Energy parameters in polypeptides. VII. Geometric parameters, partial atomic charges, nonbonded interactions, hydrogen bond interactions, and intrinsic torsional potentials for the naturally occurring amino acids». The Journal of Physical Chemistry 79 (22): 2361-2381. ISSN 0022-3654. doi:10.1021/j100589a006. Consultado el 28 de mayo de 2021.
- Arnautova, Yelena A.; Jagielska, Anna; Scheraga, Harold A. (1 de marzo de 2006). «A New Force Field (ECEPP-05) for Peptides, Proteins, and Organic Molecules». The Journal of Physical Chemistry B 110 (10): 5025-5044. ISSN 1520-6106. doi:10.1021/jp054994x. Consultado el 28 de mayo de 2021.
| Control de autoridades |
|
|---|
 Datos: Q610001
Datos: Q610001