Glioblastoma
El glioblastoma (también conocido como glioblastoma multiforme o con las siglas GBM) es el tumor más común y más maligno entre las neoplasias de la glía. Su nombre quedó establecido por la clasificación OMS-2000[2] y fijado por la clasificación OMS-2007.[3] De acuerdo con esta clasificación de la OMS de los tumores del sistema nervioso central, el nombre genérico para este tumor cerebral es "glioblastoma" y presenta dos variedades: el glioblastoma de células gigantes y el gliosarcoma.
| Glioblastoma multiforme | ||
|---|---|---|
 | ||
| Especialidad | Neurooncología | |
| Síntomas | Inicialmente inespecíficos, dolores de cabeza , cambios de personalidad, náuseas , síntomas similares a un accidente cerebrovascular | |
| Factores de riesgo | Trastornos genéticos ( neurofibromatosis , síndrome de Li-Fraumeni ), radioterapia | |
| Diagnóstico | Tomografía computarizada , resonancia magnética , biopsia de tejido[1] | |
Es un tumor de rápido crecimiento, compuesto por una mezcla heterogénea de células tumorales astrocitarias pobremente diferenciadas, con pleomorfismo, necrosis, proliferación vascular y frecuentes mitosis. Puede manifestarse a cualquier edad, pero afecta principalmente a adultos, con un pico de incidencia entre los 45 y los 70 años.[4] Se presenta habitualmente en los hemisferios cerebrales, siendo menos frecuente su localización en el tronco del encéfalo o la médula espinal. Al igual que todos los tumores cerebrales, excepto en casos muy raros, no se expande más allá de las estructuras del sistema nervioso central.[5]
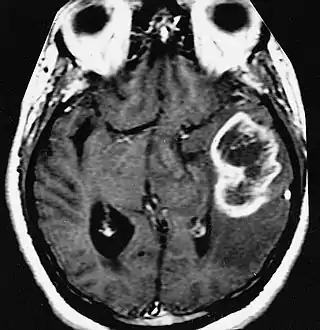
En las imágenes de TC y RM se muestra como una lesión heterogénea, de contorno irregular, que capta contraste en anillo y con un área central necrótica.
El glioblastoma puede desarrollarse a partir de un astrocitoma difuso (grado II) o de un astrocitoma anaplásico (grado III) (en tal caso se denomina secundario, ver más abajo), pero con mayor frecuencia ocurre de novo, sin ninguna evidencia de neoplasia previa (denominándose en este caso primario). Si bien el glioblastoma es el tumor cerebral primario más frecuente, su incidencia es de sólo 2-3 casos por cada 100 000 personas en Europa y Norteamérica.[6] En el tratamiento del glioblastoma intervienen la cirugía, la radioterapia y la quimioterapia. A pesar de todo el arsenal terapéutico su pronóstico es infausto, con una mediana de supervivencia de aproximadamente 14 meses.[7] Son raros los casos de supervivencia prolongada, aunque se han descrito.
Clasificación
| Tumores del sistema nervioso central |
|
Familias de tumores según la Clasificación de la OMS de 2007.
|
Clasificación OMS de 2007
En el cuadro de la derecha se relatan las familias de tumores del sistema nervioso central, de acuerdo con la clasificación de la OMS de 2007.[3]
De acuerdo con esta clasificación, en particular, el glioblastoma forma parte de los tumores astrocitarios, junto con otros seis tipos de neoplasias, conforme al siguiente esquema:
Tumores astrocitarios (Astrocytic Tumours)
- Astrocitoma pilocítico (Pilocytic Astrocytoma, IDC-O 9421/1, WHO grade I)
- Astrocitoma pilomixoide (Pilomyxoid Astrocytoma, IDC-O 9425/3, WHO grade II)
- Astrocitoma subependimario de células gigantes [Astrocitoma gigantocelular subependimario] (Subependymal Giant Cell Astrocytoma, IDC-O 9384/1, WHO grade I)
- Xantoastrocitoma pleomórfico (Pleomorphic Xanthoastrocytoma, ICD-O 9424/3,
WHO grade II) - Astrocitoma difuso [Astrocitoma] (Diffuse Astrocytoma, IDC-O 9400/3, WHO grade II)
- Astrocitoma anaplásico (Anaplastic Astrocytoma, IDC-O 9401/3, WHO grade III)
- Glioblastoma [Glioblastoma Multiforme, Astrocitoma de grado IV] (Glioblastoma, IDC-O 9440/3, WHO grade IV)
- Glioblastoma de células gigantes (Giant Cell Glioblastoma, IDC-O 9441/3, WHO grade IV)
- Gliosarcoma (Gliosarcoma, IDC-O 9442/3, WHO grade IV)
- Gliomatosis cerebri (Gliomatosis Cerebri, IDC-O 9381/3, WHO grade III)
Entre paréntesis se proporciona el nombre oficial en inglés de la Clasificación OMS-2007, junto con el código CIE-O (International Classification of Diseases for Oncology, Clasificación Internacional de Enfermedades para Oncología).[8]
A continuación se añade la gradación de la OMS bajo el lema "WHO Grade", seguida de numeración romana. Se facilita la denominación en español más utilizada en la literatura. Entre corchetes aparecen otras denominaciones. Las variantes se indican en cursiva.
Clasificación OMS de 2016
La clasificación OMS de 2016 de los tumores del sistema nervioso central[9] supone un cambio de paradigma frente anteriores clasificaciones, pues define los tumores además de por su morfología, por su perfil molecular. En concreto, el glioblastoma se clasifica según el estado de mutación del gen isocitrato deshidrogenasa (IDH): alelo-salvaje y mutante.
| IDH alelo-salvaje | IDH mutante | |
|---|---|---|
| Sinónimo | Glioblastoma primario | Glioblastoma secundario |
| Lesión precursora | Identificado de novo | Astrocitoma difuso
Astrocitoma anaplástico |
| Proporción de glioblastoma | ~90 % | ~10 % |
| Edad mediana de diagnóstico | ~62 años | ~44 años |
| Ratio hombre:mujer | 1,42:1 | 1,05:1 |
| Duración mediana de historia clínica | 4 meses | 15 meses |
| Supervivencia mediana | ||
| Cirugía + radioterapia | 9,9 meses | 24 meses |
| Cirugía + radioterapia + quimioterapia | 15 meses | 31 meses |
Historia

En la primera mitad del siglo XIX el glioblastoma se consideraba de origen mesenquimático y por tanto se definió con el término de sarcoma.[11] En 1863, Rudolf Virchow[12] demostró su origen glial. F.B. Mallory, en una memoria de 1914, propuso el término glioblastoma multiforme.[13] Sin embargo, hubo que esperar hasta 1925 para tener una descripción completa de la neoplasia, por parte de J.H. Globus e I. Strass.[14] En esta época, la denominación más común del tumor era spongioblastoma multiforme.[14] En 1926, una publicación de P. Bailey y H. Cushing volvió a proponer, con éxito, la expresión de Mallory.[15] La clasificación OMS de 2000 de los tumores del sistema nervioso fija finalmente el nombre de glioblastoma.[2] (Para una explicación histórica más detallada se remite a K.J. Zülch[16] y a D.S. Russell y L.J. Rubinstein[17]).
H.J. Scherer (1940)[18] y J.W. Kernohan et al. (1949)[19] han tenido un papel determinante en el desarrollo del concepto según el cual el glioblastoma a veces emerge por progresión y malignización de una lesión de menor grado. Este punto de vista ha recibido un fuerte apoyo por los estudios de genética molecular, que han mostrado que existe una característica acumulación secuencial de alteraciones génicas de los astrocitomas difusos de grado II al glioblastoma (véase la sección Patogénesis, Tablas 1 y 2).
Epidemiología
El glioblastoma es el tumor cerebral más frecuente,[20] representando aproximadamente el 12-15 % de todas las neoplasias intracraneales y el 50-60 % de todos los tumores astrocitarios.[16] En la mayoría de países de Europa y de América del Norte, la incidencia es de 2-3 nuevos casos al año por cada 100 000 habitantes.[6]
El glioblastoma puede manifestarse a cualquier edad, pero se presenta preferentemente en adultos, con un pico entre los 45 y los 70 años.[4] Cerca de dos tercios de los pacientes (70 %) tiene una edad comprendida en el intervalo anterior. El promedio de edad es de 53 años, con una relación varón/mujer de 1,5:1. Estas últimas cifras provienen de un estudio relativo a 1003 biopsias por glioblastoma, por parte del Hospital Universitario de Zúrich. Se citan en P. Kleihues et al. (2000).[4] Otros autores relatan cifras similares.[16] En un estudio relativo a 488 casos, G.J. Dohrman y otros[21] han demostrado que el 8,8 % de los glioblastomas son pediátricos. Los casos de glioblastomas congénitos son raros,[22] aunque los diagnósticos de gliomas malignos mediante ecografías[23][24][25] muestran que el glioblastoma prenatal puede manifestarse también a las 29 semanas de gestación.[25]
Los glioblastomas se presentan más a menudo en la materia blanca subcortical de los hemisferios cerebrales. Los sitios que más frecuentemente se ven afectados son el lóbulo temporal (31 %), el lóbulo parietal (24 %), el frontal (23 %) y el occipital (16 %). Es típica la combinación frontotemporal. La neoplasia se extiende a menudo por infiltración en la corteza adyacente, a los ganglios basales y al hemisferio contralateral. Estos datos provienen de un informe relativo a 987 glioblastomas, realizado por el Hospital Universitario de Zúrich. Se citan en P. Kleihues et al. (2000).[4] Los glioblastomas intraventriculares son excepcionales.[26] Los glioblastomas del tronco encefálico son poco frecuentes, si bien en niños su incidencia es significativa.[21] El cerebelo y la columna vertebral raramente resultan afectados por esta neoplasia.[4]
Etiología
Visión tradicional
Los tumores se forman como resultado de un crecimiento anormal y no regulado de las células. Así, las células tumorales presentes en el cerebro retoman el "ciclo celular", debido a las alteraciones en algunos de los muchos genes que controlan la división celular y el crecimiento. Aunque se sabe mucho sobre las alteraciones de estos genes en los tumores cerebrales, la principal razón por la cual surgen estas alteraciones es, de hecho, desconocida en la actualidad.[27]
Heredabilidad
Téngase en cuenta que cuando se habla de genes, no quiere decirse que los tumores cerebrales sean hereditarios. Si bien existen síndromes en los que estos tumores presentan familiaridad, estas situaciones (neurofibromatosis, síndrome de Turcot, síndrome de Li-Fraumeni, etc.) son muy poco frecuentes y normalmente conocidas por la familia antes de que se desarrolle un tumor en un miembro familiar.[27]
Factores de riesgo
A continuación se hará hincapié en los posibles factores desencadenantes identificados hasta el momento.
La radiación ionizante es el único factor de riesgo inequívoco que se ha identificado para los tumores gliales y meníngeos. La irradiación del cráneo, incluso a dosis bajas, puede aumentar la incidencia de los tumores gliales en un factor de 3 a 7 y de los meningiomas en un factor de 10, con un período de latencia de 10 a más de 20 años después de la exposición.[28][29] Ninguna otra situación ambiental o de comportamiento del paciente ha sido identificada claramente como un factor de riesgo. Se ha informado de que el uso de teléfonos móviles y tintes para el cabello, la proximidad de cables de alta tensión, trauma craneal, dietas con N-nitrosaminas, u otros factores nutricionales, aumentan el riesgo de tumores cerebrales.[30][31][32][33] Sin embargo, estos datos se consideran discutidos y no convincentes.[34]
La asociación entre el tipo de ocupación profesional y la aparición de glioblastomas ha sido objeto de numerosos estudios. Los trabajadores expuestos continuamente a cloruro de vinilo, compuestos con base fenólica e hidrocarburos aromáticos resultaron ser los de mayor riesgo.[35][36][37][38][39]
Células madre neoplásicas del cerebro
Ya en los años sesenta y sobre todo a partir de los años noventa, estudios realizados primero en animales y posteriormente en humanos han demostrado que en el interior del cerebro hay una producción continua de nuevas células. Particularmente en la zona subgranular del giro dentado del hipocampo y en la zona subventricular de los ventrículos laterales se han identificado células madre neurales capaces de producir progenitores neurales que darán lugar a neuroblastos o neuronas inmaduras que luego se diferenciarán a neuronas (interneuronas). Estas células madre neurales, si son aisladas y cultivadas in vitro con los medios de cultivo y protocolos adecuados, también pueden diferenciarse a astrocitos u oligodendrocitos.[40][41][42][43][44][45] También son capaces de autorrenovarse, permitiendo por tanto que el número total de células se mantenga constante.[46]
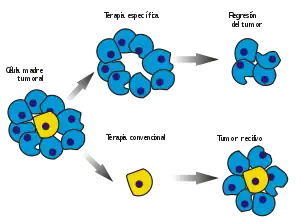
Por otro lado, una línea de investigación ha descubierto, a partir de 2002, que en los tumores cerebrales, en particular en los glioblastomas, existe una jerarquía de células tumorales, en el sentido de que una parte (pequeña) del tumor está compuesta por células que tienen las mismas características que las células madre neuronales, por lo que los autores han acuñado el nombre de células madre neoplásicas del cerebro (brain tumor stem cells).[47][48][49]
Son éstas el motor del tumor: reproducen continuamente células madre tumorales y células tumorales (no madre). Y son sólo las últimas en ser objeto de los ataques de las terapias. Las células madre neoplásicas son en efecto resistentes a la radioterapia y la quimioterapia, pues son capaces de auto-reparar a tiempo los daños efectuados por las terapias tradicionales, antes de que éstos se conviertan en irreversibles y capaces de desactivar la célula.[50][51] [52] [53] [54]
Por tanto, basta con que sólo una célula madre neoplásica cerebral escape de la cirugía para que se ponga en movimiento el mecanismo y se produzca una recidiva. Se supone que la existencia de estas células madre neoplásicas del cerebro se debe a un error en la autorregulación de las células madre neuronales (autorrenovación), mencionada anteriormente.[46]
El marco conceptual que se expone aquí de manera muy sucinta, en la literatura se denomina "hipótesis de las células madre tumorales (o cancerosas)". Este marco es el que sigue la inmensa mayoría de investigadores. Sin embargo, existe una minoría pequeña, pero notoria que tiende a dar una explicación diferente de los fenómenos descritos o a ponerlos en un marco conceptual diferente.[55][56] [57][58] [59]
Patogénesis
A continuación se expone (mediante tablas) la secuencia de alteraciones genéticas que conducen al glioblastoma, tal como se describe en las dos últimas ediciones de la clasificación de la OMS de los tumores del sistema nervioso central.[4] [60] Se distinguen dos tipos de alteraciones:
- la activación de factores oncogénicos:
- EGF/R (Epidermal Growth Factor/Receptor, Factor de crecimiento epidérmico)
- MDM2 (la oncoproteína Mouse Double Minute 2 promueve la supervivencia celular y la progresión del ciclo celular mediante la inhibición del supresor tumoral TP53[61])
- PDGF/R (Platelet-Derived Growth Factor/Receptor, Factor de crecimiento derivado de plaquetas)
- la desactivación de factores oncosupresores:
- 10p, 10q, 19q (Cromosomas)
- DCC (Deleted in Colorectal Cancer tumor suppressor gene, Gen con deleción en el cáncer colorrectal)
- p16 (Tumor suppressor gene/protein, Antígeno supresor tumoral)
- TP53 (Tumor suppressor gene/protein, Antígeno supresor tumoral)
- PTEN (Phosphatase and TENsin homolog es un supresor tumoral que controla el crecimiento, la proliferación y la supervivencia celular. De su mutación o inhibición puede desencadenarse la aparición de tumores, por ejemplo, de próstata, mama, colon y cerebro.[62][63][64])
- RB (RetinoBlastoma tumor suppressor gene, Proteína del retinoblastoma)
La Tabla 1 (P. Kleihues y H. Ohgaki, 1999,[65] como aparece en P. Kleihues et al., 2000,[4] con modificaciones gráficas) se ha tomado de la clasificación de la OMS de 2000 y muestra las mutaciones que se producen desde de las células sanas hasta el glioblastoma.
A la izquierda se puede ver la activación de las lesiones intermedias (astrocitoma difuso y astrocitoma anaplásico) antes de llegar al glioblastoma llamado secundario (aquí este adjetivo no tiene normalmente el significado de metastásico sino de derivado de lesiones anteriores). En la parte derecha de la tabla se muestran las mutaciones que de las células sanas conducen directamente (de novo) al glioblastoma, llamado por tanto primario.
(Entre paréntesis figura el porcentaje de presencia de la alteración sola).
| Tabla 1. Alteraciones genéticas del Glioblastoma (2000)[4] | |
| Células astrocíticas diferenciadas o precursores neuroepiteliales | |
| Mutación de TP53 (>65 %) Sobreexpresión de PDGF-A, PDGFR-α (~60 %) ↓ | EGFR: Amplificación (~40 %) Sobreexpresión (~60 %) MDM2: Amplificación (< 10 %) Sobreexpresión (~50 %) Deleción de p16 (30-40 %) Pérdida de heterocigosis en 10p y 10q Mutación de PTEN (~30 %) Alteración de RB ↓ |
| Astrocitoma difuso | |
| Pérdida de heterocigosis en 19q (~50 %) Alteración de RB (~25 %) ↓ | |
| Astrocitoma anaplásico | |
| Pérdida de heterocigosis en 10q Mutación de PTEN (5 %) Pérdida de expresión de DCC (~50 %) Amplificación de PDGFR-α (< 10 %) ↓ | |
| Glioblastoma secundario | Glioblastoma de novo |
La Tabla 2 (P. Kleihues et al., 2007,[60] con algunas modificaciones gráficas) proviene de la clasificación de la OMS de 2007. Se resumen aquí años posteriores de estudio y profundización.[66] [67]
Nótese que el asterisco (*) indica alteraciones genéticas que difieren significativamente en frecuencia entre los glioblastomas primarios y secundarios.
| Tabla 2. Alteraciones genéticas del Glioblastoma (2007)[60] | ||
| Células astrocíticas diferenciadas, o precursores o madre | ||
↓ | ↓ | I I I ↓ <3 meses (68 %) <6 meses (84 %) I I I I ↓ |
| Astrocitoma difuso | ||
↓ | I ↓ 5,1 años I I ↓ | |
| Astrocitoma anaplásico | ||
1,9 años ↓ | ||
| Pérdida de heterocigosis en 10q (63 %) Amplificación de EGFR (8 %) Deleción de (19 %) Mutación de TP53 (65 %)* Mutación de PTEN (4 %) ↓ | Amplificación de EGFR (36 %)* Deleción de (31 %) Mutación de TP53 (28 %) Mutación de PTEN (25 %)* ↓ | |
| Glioblastoma secundario | ||
| 5 % de los casos Edad media: 45 años Proporción H/M: 0,65 | Edad media: 62 años Proporción H/M: 1,33 | |

Las relaciones entre las dos tablas pueden deducirse examinando las referencias bibliográficas indicadas. No obstante, es necesario destacar un hecho. Un examen, incluso superficial, de estas tablas lleva a la conclusión de que los glioblastomas primarios y secundarios son dos enfermedades distintas (aunque poco distinguibles histológicamente), que afectan a diferentes grupos de pacientes por edad y sexo y se desarrollan a través de diferentes vías genéticas, con diferentes perfiles de expresión proteica y ARNm. Estas diferencias son importantes, sobre todo porque pueden influir en la respuesta del tumor a radioterapia y quimioterapia y pueden ser objeto de futuros enfoques terapéuticos.[67]
Complicaciones
En el siguiente esquema se muestra un resumen de las complicaciones ligadas al glioblastoma, en el que se distinguen las debidas a la enfermedad y las relacionadas más estrictamente con el tratamiento.[68] Muchas de estas complicaciones no son comunes y un número significativo de ellas pueden controlarse terapéuticamente de manera eficaz.
- Complicaciones relacionadas con el tumor:
- Edema
- Trastornos neurológicos
- Trastornos visuales
- Hidrocefalia
- Gliomatosis leptomeníngea
- Deterioro de las funciones cognitivas
- Deterioro del estado psicológico (ansiedad, etc.)
- Complicaciones relacionadas con el tratamiento:
- Patologías relacionadas con la cirugía
- Infecciones
- Trastornos neurológicos
- Trastornos visuales
- Trastornos relacionados con la radioterapia
- Trastornos neurológicos
- Trastornos visuales
- Deterioro de las funciones cognitivas
- Trastornos relacionados con la quimioterapia
- Disfunciones hemáticas
- Trastornos del aparato respiratorio
- Diarrea
- Fatiga
- Trastornos neurológicos
- Disfunciones hemáticas
- Trastornos relacionados con los fármacos anticonvulsivantes
- Trastornos relacionados con los fármacos antiinflamatorios
- Trastornos relacionados con los fármacos citostáticos
Anatomía patológica
Examen macroscópico
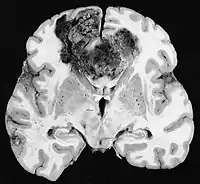
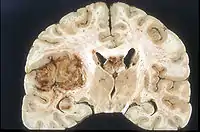
A pesar de la corta duración de los síntomas, los glioblastomas suelen ser grandes en el momento de la presentación y pueden ocupar más de un lóbulo. La lesión es en general unilateral, aunque las del tronco cerebral y el cuerpo calloso pueden tener simetría bilateral. El tumor ocupa la misma posición en los dos hemisferios y muestra un aspecto de "mariposa". La extensión supratentorial bilateral se debe a un rápido crecimiento a lo largo de las estructuras mielinizadas, en particular a través del cuerpo calloso y a lo largo del fórnix hacia los lóbulos temporales. Los límites de la masa neoplásica, que no está encapsulada, son difusos por todas partes. La coloración es grisácea, pero pueden encontrarse abundantes variaciones de color, causadas por necrosis o hemorragias más o menos recientes, por lo que sobre el fondo gris aparecen zonas amarillentas, por degeneración grasa o necrosis y zonas de color rojizo o negruzco debidas a hemorragia.
La zona periférica del tejido tumoral hipercelular aparece como un borde suave y gris. El tejido necrótico puede bordear estructuras cerebrales adyacentes sin una zona tumoral intermedia detectable macroscópicamente. La necrosis central puede ocupar más del 80 % de la masa total del tumor.
Los glioblastomas están generalmente salpicados de manchas rojas y marrones debidas al sangrado. A veces son lo suficientemente grandes como para causar síntomas similares a un accidente cerebrovascular, que puede ser el primer signo clínico del tumor. Los quistes macroscópicos, cuando están presentes, contienen un fluido turbio proveniente del tejido tumoral necrótico licuado, en claro contraste con los quistes de retención bien definidos de los astrocitomas difusos de grado II.
La mayoría de los glioblastomas de los hemisferios cerebrales son claramente intraparenquimatosos, con epicentro en la materia blanca. A veces la neoplasia se presenta como ampliamente superficial y en contacto con leptomeninge y duramadre, y se puede confundir con un carcinoma metastásico o con una lesión extra-axial, como el meningioma.[11][60]
Examen microscópico
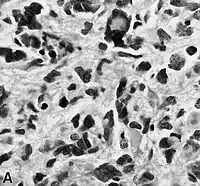
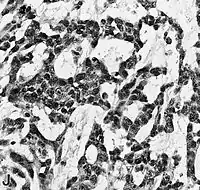
El glioblastoma es una neoplasia anaplásica de la glía compuesta por células tumorales astrocíticas pobremente diferenciadas, polimórficas, con marcadas atipias nucleares y una intensa actividad mitótica. Otras características peculiares a fines diagnósticos son también la marcada proliferación microvascular y la presencia de necrosis. Como sugiere el adjetivo "multiforme", la morfología histológica del glioblastoma es extremadamente variable, con células redondeadas, en forma de huso, de dimensiones más bien pequeñas o muy grandes. Mientras que algunos glioblastomas muestran un alto grado de polimorfismo celular y nuclear, con numerosas células gigantes plurinucleadas, otros presentan una conformación caracterizada por una celularidad intensa, pero más bien repetitiva.
La naturaleza astrocítica de la neoplasia puede resultar bastante fácil de identificar, al menos localmente, en algunos tumores, pero difícil de reconocer en otros, debido al alto grado de anaplasia. La heterogeneidad de región a región del glioblastoma es relevante y dificulta el diagnóstico en muestras limitadas, como las obtenidas por biopsia estereotáxica[69] (véase la fotografía en Cirugía). A pesar de la presencia predominante de células poco diferenciadas, en algunos puntos se pueden distinguir astrocitos neoplásicos más diferenciados. Esto es particularmente cierto en los casos de glioblastoma como resultado de la progresión de un astrocitoma difuso (grado II de la escala OMS). La transición entre zonas que aún poseen diferenciación astrocítica reconocible y zonas de alta anaplasia celular puede ser continua o repentina. Un cambio brusco en la morfología refleja habitualmente la aparición de un tumor diferente, fruto de la adquisición de una o más alteraciones genéticas adicionales.[70]
En el contexto de la neoplasia se observan grandes áreas de necrosis, rodeadas de núcleos dispuestos paralelos entre sí, formando "empalizadas" típicas. Se encuentra una marcada proliferación de células endoteliales con formación de numerosos vasos, a veces con apariencia de montón o madeja. Algunos tienen pared hialina y otros están trombosados. La proliferación endotelial, sin embargo, no es difusa sino focalizada en algunos puntos. Alrededor de la neoplasia se pueden encontrar zonas de astrocitos gemistocíticos (astrocitomas difusos de grado II).[11][60]
Clínica
Signos y síntomas
La historia clínica de la enfermedad suele ser corta (menos de 3 meses, en más del 50 % de los casos), a menos que el tumor no se desarrolle por la progresión de un astrocitoma de bajo grado (glioblastoma secundario).
Los síntomas del glioblastoma son los de una masa expansiva en el interior del cráneo, que aumenta la presión intracraneal. Es común por tanto encontrar cefalea, náuseas, vómitos, dilatación de los vasos cerebrales con alteraciones de la retina hasta el papiledema, hemiparesia, hemianestesia, hemianopsia, diplopía, afasia y crisis convulsivas. El porcentaje de los pacientes que experimentan ataques epilépticos asciende a un tercio.
Destacan también síntomas neurológicos no específicos tales como el obnubilamiento de la conciencia y los cambios de personalidad.[60]
Diagnóstico por imagen y tumores cerebrales
La presencia de un tumor cerebral puede ser revelada con eficacia a través de la tomografía computarizada (TC) y la resonancia magnética (RM).
La RM tiene una sensibilidad mayor en comparación con la TC en la identificación de lesiones, pero no siempre es de fácil acceso para el paciente y presenta algunos inconvenientes: no se puede hacer a pacientes con marcapasos, prótesis incompatibles con el campo magnético, clips metálicos, etc. La TC sigue siendo el método de elección en la detección de calcificaciones internas a las lesiones o de erosiones óseas de la base del cráneo.
El uso de un medio de contraste (yodo en el caso de TC, paramagnético en el caso de RM - gadolinio), permite obtener información sobre la vascularización y sobre la integridad de la barrera hematoencefálica, una mejor definición del nódulo tumoral respecto al edema circundante y permite avanzar hipótesis sobre el grado de malignidad.
El examen radiológico permite también evaluar los efectos mecánicos (y los consiguientes cambios en la relación de las estructuras cerebrales) derivados de la presencia de la masa "extraña": hidrocefalia y hernias, cuyos efectos también pueden ser letales.
El examen, en vistas a la operación quirúrgica, precisa la localización de la lesión y la proximidad (o incluso la implicación) del tumor a áreas cerebrales absolutamente vitales (áreas conocidas como "elocuentes"). En este sentido, la RM resulta superior a la TC por el hecho de que es capaz de proporcionar imágenes tridimensionales.[71]
Antes de cerrar esta sección, conviene llamar la atención sobre algunos conceptos y términos que serán de utilidad para la comprensión de las siguientes secciones.
Aspecto radiológico del tejido neoplásico
Se pretende poner de relieve el fenómeno de alteración desde el punto de vista radiológico del tejido neoplásico respecto al parénquima cerebral normal (modificaciones de la densidad electrónica de los materiales en el caso de la TC y de la intensidad de señal en la RM).
Al igual que la mayoría de los tejidos patológicos, los tumores se caracterizan por una mayor acumulación de agua intracelular. Aparecen hipodensos en la TC, es decir, de densidad inferior a la del parénquima cerebral. En la RM aparecen hipointensos en las imágenes ponderadas en T1 e hiperintensos en las ponderadas en DP y T2.[38][72]
Realce de contraste (aumento de la señal del contraste)
En una placa radiográfica, la zona de cerebro sano, no debería mostrar luminiscencias específicas. Por tanto, es natural que se preste atención a las porciones de mayor señal de contraste.
En el tumor, en general, la mayor parte del "realce de contraste" se debe a la especial barrera hematotumoral, que permite el paso de yodo (TC) y gadolinio (RM) al espacio intersticial extravascular intratumoral: aumenta por tanto la señal (densidad o intensidad) del tumor.
No obstante, hay que tener en cuenta que el "realce de contraste" no delimita con certeza el tumor del edema perilesional, de hecho, en los gliomas infiltrantes malignos (como por ejemplo, el glioblastoma y astrocitoma anaplásico) la muestra anatomopatológica refleja tejido neoplásico incluso más allá del edema vasogénico (causado por la destrucción de la barrera hematoencefálica por parte del tumor), algo que no es fácilmente demostrable por medio de imágenes radiográficas. [38][72]
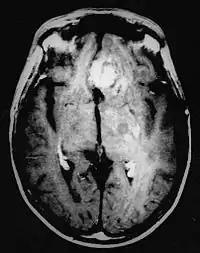
Control posquirúrgico
El control posquirúrgico mediante RM (o TC) para la determinación de la radicalidad de la remoción de un tumor se considera cuestionable en la literatura: el examen se debería realizar dentro de 24 horas después de la cirugía, es decir, antes de que se instauren las alteraciones de la barrera hematoencefálica apoyadas por fenómenos fibrótico-cicatriciales; en otras palabras, la cicatriz fisiológica tiene un realce de contraste que puede confundirse fácilmente con un residuo o con una recidiva del tumor.[73]
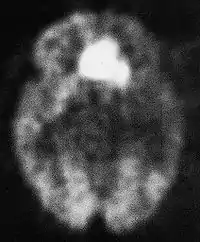
Incluso después del tratamiento radioquirúrgico, una radionecrosis (véase más abajo la sección homónima) puede presentar características de captación de contraste con aspecto similar al de un glioma maligno. Solo por medio de métodos funcionales, tales como la tomografía por emisión de positrones (PET) con fluorodesoxiglucosa (FDG-PET), la cual demuestra una mayor captación de glucosa por parte del tumor respecto del tejido sano, es posible evaluar la ausencia de metabolismo en las necrosis respecto a la recurrencia tumoral (aunque es posible que necrosis y recurrencia coexistan).[73] Como alternativa a la PET se puede utilizar el análisis espectroscópico mediante RM: en los "mapas" metabólicos de este método está presente el pico de colina (Cho), que se asocia con la síntesis de membranas celulares; otro pico es indicativo del elevado turnover celular, como ocurre en los tumores.[38][72]
Diagnóstico por imagen y glioblastoma
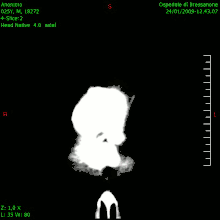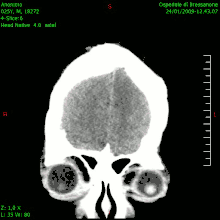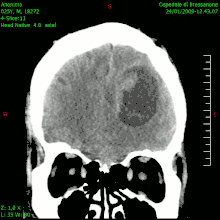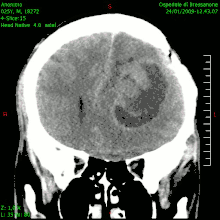
La TC muestra una lesión de morfología irregular, predominantemente hipodensa y fuertemente dishomogénea, debido a la presencia de grandes áreas necróticas de más clara hipodensidad y de áreas sólidas hiperdensas. Estas últimas son la expresión de un rápido crecimiento y por lo tanto de una elevada malignidad. Son frecuentes las zonas hemorrágicas, que van desde pequeños focos a grandes áreas hemáticas que pueden cubrir toda la lesión. Es característica la morfología en "mariposa" si el tumor se asienta en ambos hemisferios a través del cuerpo calloso.
Tras la aplicación de contraste aparecen gruesos anillos alrededor de las áreas necróticas. En la RM, la parte sólida aparece hipointensa en T1 e hiperintensa en T2 con zonas de señal más elevada en las partes de mayor celularidad. Las áreas necróticas, hiperintensas en T2, pueden presentarse hipo-, iso- o hiperintensas en T1 en función del contenido proteico o de productos de la degradación de la hemoglobina. El realce tras el contraste suele ser intenso e irregular en la periferia del tumor e identifica sobre todo la componente celular "proliferativa" de la neoplasia. Son comunes las áreas puntiformes y serpiginosas de ausencia de señal de flujo, asociadas a la presencia de una neovascularización rica. Estos vasos de neoformación patológica carecen de barrera hematoencefálica, lo que explica tanto la abundante impregnación como el edema vasogénico perilesional (véase la sección anterior), debido al paso de líquido al medio extracelular.[71][72]
Diagnóstico diferencial
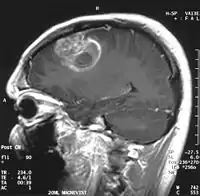

El diagnóstico diferencial se hace con: metástasis, hemorragia cerebral espontánea, abscesos, formas atípicas de esclerosis múltiple o linfoma.[74]
Diagnóstico por imagen. Conclusiones
Podemos concluir afirmando que el primer paso que hay que considerar en la evaluación de un paciente en el que se sospecha una neoplasia cerebral es la resonancia magnética. Dicho examen también se recomienda a los pacientes aquejados de crisis epilépticas para las que no se encuentra una justificación inmediata y plausible.
Normalmente, la resonancia revela la presencia del glioblastoma sin mayor dificultad como causa de los síntomas referidos y no se necesitan pruebas posteriores.[75]
Tratamiento
El glioblastoma resulta muy difícil de tratar, debido a varios factores:[76][77]
- Las células tumorales son muy resistentes a los tratamientos convencionales;
- El cerebro es susceptible de sufrir daños debidos a estos tratamientos;
- El cerebro tiene una capacidad muy limitada para repararse a sí mismo;
- Muchos fármacos no pueden atravesar la barrera hematoencefálica para actuar sobre el tumor.
En el tratamiento del glioblastoma, como en cualquier otro tumor cerebral, distinguimos entre tratamientos de apoyo y tratamientos curativos.[78][79][80]
Tratamiento de apoyo
El tratamiento de apoyo tiene como objetivo paliar los síntomas y mejorar las funciones neurológicas del paciente. Los agentes de apoyo primarios son los fármacos antiepilépticos y los corticosteroides.
Fármacos antiepilépticos
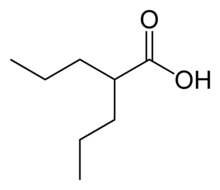
Los fármacos antiepilépticos se administran a un 25 % de los pacientes que han tenido crisis epilépticas en la presentación de la enfermedad. La fenitoína (300-400 mg/d) es el fármaco más utilizado, pero la carbamazepina (600-1000 mg/d), el fenobarbital (90-150 mg/d) y el ácido valproico (750-1500 mg/d), son igualmente eficaces. Las dosis de estos anticonvulsivantes deben adaptarse a los niveles séricos adecuados para proporcionar la máxima protección.
Igualmente efectivos son los antiepilépticos de reciente desarrollo, como el levetiracetam, la gabapentina, la lamotrigina y el topiramato. La mayoría de estos nuevos principios activos tienen la ventaja de causar menos efectos secundarios de tipo cognitivo y no alteran el metabolismo de los agentes quimioterápicos porque no inducen el sistema microsomal hepático. Estos nuevos anticonvulsivantes están reemplazando rápidamente a los fármacos tradicionales en el tratamiento antiepiléptico de primera línea.[75]
Profilaxis
Los ensayos clínicos prospectivos han dado resultados negativos en el intento de demostrar la eficacia de un uso profiláctico de los fármacos antiepilépticos en el caso de pacientes con tumores cerebrales que nunca habían padecido crisis epilépticas. En consecuencia, la literatura médica no recomienda su uso para este propósito, excepto para el período perioperatorio, cuando su uso puede reducir la incidencia de crisis epilépticas posoperatorias.
En el caso de pacientes que nunca han experimentado crisis, se recomienda reducir gradualmente el suministro de anticonvulsivantes a las dos semanas de la intervención.[75][78][81]
Corticosteroides

Los fármacos a base de corticosteroides pueden reducir el edema peritumoral, disminuyendo el efecto masa de la neoplasia y reduciendo la presión intracraneal. Como efecto inmediato se observa un alivio de los dolores de cabeza y una mejora de los signos lateralizantes.
El corticosteroide de elección es la dexametasona, debido a su mínima actividad mineralocorticoide. La dosis inicial es de aproximadamente 16 mg/d. Esta cantidad puede ser aumentada o disminuida hasta alcanzar la dosis mínima necesaria para controlar los síntomas neurológicos. El uso prolongado de corticosteroides se asocia con hipertensión, diabetes mellitus, estado hiperglucémico hiperosmolar no cetósico, miopatía, aumento de peso, insomnio y osteoporosis. Así, en el paciente con tumor cerebral la dosis de esteroides debe ser reducida gradualmente "tan pronto como sea posible", una vez que haya comenzado el tratamiento curativo. Para la mayoría de los pacientes la administración de corticosteroides puede detenerse una vez hayan completado la radioterapia.
En el caso de pacientes tratados con corticosteroides durante más de 6 semanas se recomienda la profilaxis antibiótica para la neumonía por Pneumocystis carinii. Este tratamiento debe continuar durante 1 mes después de la retirada de los corticosteroides.[75]
Tratamiento curativo
El tratamiento curativo de los tumores cerebrales comprende principalmente la cirugía, la radioterapia y la quimioterapia.
El primer paso es, si es posible, la elaboración de un plan general de tratamiento que permita trazar la secuencia y los elementos individuales del tratamiento multidisciplinario.
Cirugía
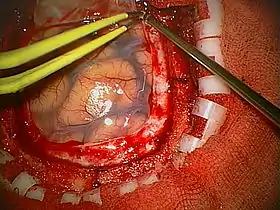

El abordaje quirúrgico debe elegirse cuidadosamente con el fin de lograr la máxima resección posible del tumor, sin afectar a las estructuras vitales del cerebro y reduciendo al mínimo el riesgo de déficit neurológico posoperatorio.
Los objetivos de la intervención quirúrgica son:
- Obtener un diagnóstico histológico preciso;
- Reducir el efecto masa causado por el tumor y/o el edema peritumoral;
- En su caso, mantener o restablecer el flujo del líquido cefalorraquídeo;
- Conseguir una (potencial) curación a través de la remoción "total" de la neoplasia (en el caso del glioblastoma, es muy raro que la cirugía logre la curación, aun así, puede reducir el tamaño del tumor con el fin de hacerlo más manejable para la radio- y la quimioterapia).
Una resección superior al 98 % del volumen del tumor (resección "total") aumenta la supervivencia en comparación con una resección parcial o subtotal. La resección subtotal "extensa" no parece proporcionar ninguna ventaja de supervivencia en comparación con la biopsia o la resección parcial.[75][82]
A partir de 2006, se demostró la utilidad de utilizar cirugía guiada con fluorescencia de 5-aminolevulínico para mejorar la visualización y el grado de extirpación de estos tumores. Desde esa fecha hasta 2019, se ha generalizado su uso en casi todo el mundo, confirmándose que la tasa de extirpación es superior a la técnica convencional.[83]
En caso de recurrencia de la enfermedad (que ocurre en casi todos los glioblastomas), de expansión de la parte del tumor remanente de la operación quirúrgica, o de radionecrosis (tanto la recurrencia de la enfermedad como la radionecrosis causan efecto masa y edema y, como se ha mencionado anteriormente, no son distinguibles en la resonancia clásica) se recurre a una segunda intervención, para reducir el efecto de la masa neoformada en el parénquima cerebral. En situaciones de recurrencia es difícil alcanzar la curación. No obstante, sí suele conseguirse una mejora en la calidad de vida y un modesto alargamiento de la mediana de supervivencia.
En general, está excluida una segunda operación en los pacientes con un índice de Karnofsky (KPS) menor o igual a 60, o en aquellos pacientes que no son candidatos a tratamientos adyuvantes sucesivos a la cirugía.[75][82] Antes de concluir esta sección, merece la pena hacer referencia a estudios clínicos que implican, durante la intervención quirúrgica, la administración intratecal de quimioterápicos, inmunoterápicos o líquidos radiactivos. Estos estudios están en primera fase de experimentación.[84][85][86] El posicionamiento en la mesa de operaciones de "wafers" impregnados con carmustina es el único caso de quimioterapia intracavitaria aprobado en la actualidad por la FDA (Food and Drug Administration) para glioblastomas.[87][88]
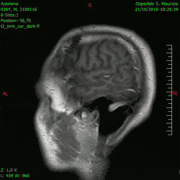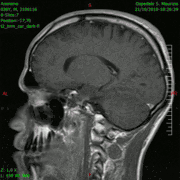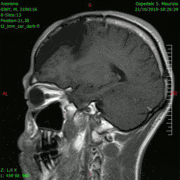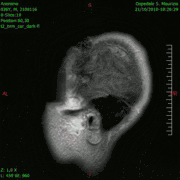 Animación en el plano sagital de un caso de resección total gruesa de un glioblastoma
Animación en el plano sagital de un caso de resección total gruesa de un glioblastoma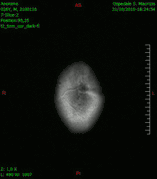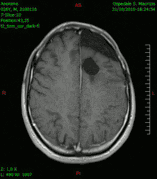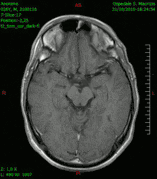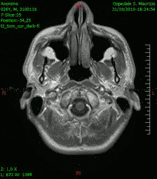 Animación en el plano axial
Animación en el plano axial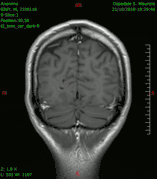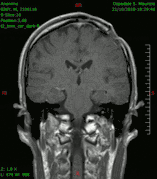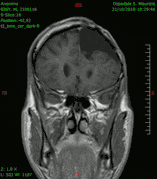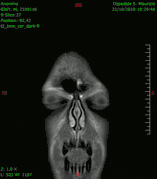 Animación en el plano coronal
Animación en el plano coronal
Radioterapia
La radioterapia, que se realiza generalmente después de la intervención quirúrgica, se aplica a la parte del encéfalo afectada por la intervención, así como a un cierto margen externo. Su objetivo es dañar el ADN de las posibles células tumorales que hayan podido permanecer después de la cirugía, ya que no son visibles al microscopio (por ser infiltrantes, están más o menos alejadas de la zona de operación). Si la terapia de radiación consigue dañar estas células antes de que tengan la capacidad de reparar el ADN y continuar la multiplicación celular, el paciente gana en supervivencia.
Los ensayos clínicos en gliomas de alto grado (astrocitoma anaplásico, oligodendroglioma anaplásico, oligoastrocitoma anaplásico, glioblastoma) efectuados por el BTSG (Brain Tumor Study Group) han mostrado que la radioterapia posoperatoria a dosis superiores a 50 Gy proporciona una mejora en la supervivencia respecto a ningún tratamiento posoperatorio, y que 60 Gy dan como resultado una supervivencia significativamente mayor en comparación con 50 Gy.[89][90]
Esta cantidad de radiación corresponde a una dosis apenas superior a la necesaria para la formación de radionecrosis, por lo que se ha elegido como estándar el tratamiento radioterápico de 60 Gy, administrados en un total de 30-33 fracciones, una al día.[75][90][91] Pacientes con glioblastoma de más de 60 años con una terapia acortada de 40 Gy en 15 fracciones mostraron una supervivencia idéntica a la obtenida con el tratamiento estándar. Por tanto, para estos pacientes es razonable el uso de este tratamiento reducido.[92]
Alrededor de la mitad de los pacientes con astrocitoma anaplásico "responden" a la radioterapia con 60 Gy. El porcentaje desciende al 25 % para los pacientes con glioblastoma. En ambas neoplasias, los casos de curación completa por radioterapia son muy raros.[75]
En un esfuerzo por mejorar los resultados mencionados, se han desarrollado una serie de nuevos enfoques como la radioterapia hiperfraccionada (HFRT), la braquiterapia (uso de agujas radiactivas depositadas directamente) o la radiocirugía. Esta última ha tenido en el pasado reciente un cierto interés, ya que es un procedimiento no invasivo que se puede realizar en varias situaciones, incluso en hospitales de día. Requiere una selección muy cuidadosa de los pacientes, porque, entre otras cosas, requiere que la neoplasia no sea extensa, sino altamente focalizada. Salvo en casos especiales, estas nuevas técnicas no han mostrado una mejora significativa de la supervivencia global de los pacientes.[75][90]
Radionecrosis
Ya hemos mencionado en apartados anteriores la necrosis radioinducida. Esta complicación se produce principalmente por la braquiterapia y la radiocirugía, y determina la sintomatología por efecto masa, descrita anteriormente, en cerca del 50 % de los pacientes con un glioma maligno. Con el tratamiento con corticosteroides a menudo es posible controlar el edema circundante al área radionecrótica. Esto, a su vez, produce a la larga dependencia a los esteroides, con todas las complicaciones por uso prolongado que ello conlleva (véase la sección de corticosteroides). En los casos más graves es necesario recurrir a la cirugía para extirpar la masa necrótica.[75]
Quimioterapia
La quimioterapia tiene la misión de dañar la organización del ADN de las células tumorales que puedan permanecer después de la cirugía o haber escapado de la radiación. Si el agente quimioterápico es capaz de alterar ese ADN, la célula tumoral pasa a una fase de "muerte programada" (apoptosis).

La quimioterapia ofrece beneficios limitados para los pacientes de glioblastoma. En los ensayos clínicos, el uso de nitrosoureas no ha ampliado significativamente la mediana de supervivencia en todos los pacientes, pero un subgrupo de ellos parece beneficiarse de una prolongación de la supervivencia con la adición de quimioterapia a la radioterapia. Los factores pronósticos como la edad, el índice de Karnofsky, etc., no pueden predecir qué pacientes se beneficiarán de la quimioterapia.[75]
En un gran estudio de fase III, los pacientes (diagnosticados de glioblastoma y sin tratamiento radio-quimioterápico precedente) fueron "aleatorizados" para recibir radioterapia sola (grupo A) o radioterapia con administración concomitante diaria de temozolomida, seguida de administración adyuvante mensual de temozolomida (grupo B). De un total de 573 pacientes, la supervivencia media aumentó de 12,1 meses (grupo A) a 14,6 meses (grupo B). Pero lo más significativo fue que la supervivencia a más de 2 años pasó a ser más del doble, pasando del 10,4 % del grupo A al 26,5 % del grupo B.[93]
El tratamiento combinado de radioterapia y temozolomida resultó medianamente bien tolerado y con una toxicidad adicional mínima, por lo que este protocolo se ha convertido en el tratamiento estándar de elección para todos los nuevos pacientes de glioblastoma.[75] Como producto colateral del estudio mencionado anteriormente, se ha identificado una proteína tumoral (MGMT) que puede predecir, con una aproximación útil en la práctica, qué pacientes se beneficiarán del protocolo combinado. Este método todavía está en fase de pruebas por parte de la comunidad científica y sólo se cita aquí a título informativo.[94][95][96]
Cannabinoides
Mención aparte merecen los cannabinoides. Se sabe que los derivados del cannabis son eficaces en oncología (a través de cápsulas de tetrahidrocannabinol (THC) o de su análoga sintética nabilona), por un lado, para combatir las náuseas y los vómitos inducidos por la quimioterapia, y por otro, para estimular el apetito y disminuir la sensación de angustia o el dolor en sí.[97][98] Está demostrada su capacidad para inhibir el crecimiento y la angiogénesis en los gliomas malignos.[99][100] Los resultados de un estudio piloto sobre el uso de THC en pacientes (en fase terminal) con glioblastoma recurrente han resultado ser dignos de profundización.[101]
Pero lo más interesante es el descubrimiento (por ahora confirmado en animales) de que los cannabinoides son capaces de atacar a las células madre neoplásicas del glioblastoma, con el resultado, por un lado, de inducir su diferenciación en células más maduras (y por lo tanto más "tratables") y por otro lado de inhibir la tumorigénesis.[102]
Otros tratamientos
Desafortunadamente, la mediana de supervivencia con cirugía, quimioterapia y radioterapia, es de unos 14,7 meses. Por eso se buscan otras estrategias terapéuticas que puedan ser eficaces, sin los efectos secundarios tan importantes de la quimioterapia. Entre las más prometedoras ahora mismo, están la inmunoterapia, y los virus oncolíticos. Por ejemplo con células dendríticas, los resultados provisionales de un estudio fase 2 realizado en España, muestran una mediana de supervivencia de 27,4 meses.[103] Con virus oncolíticos, se ha diseñado un virus capaz de destruir células tumorales sí y a las sanas no, un primer estudio en USA ha dado resultados prometedores, y se está iniciando otro en España.[104]
Recurrencia
A pesar del éxito inicial (limitado) del tratamiento, prácticamente todos los glioblastomas se reproducen.[105]
En esta situación, el paciente puede someterse a una segunda intervención (si se dan las condiciones adecuadas) o puede beneficiarse de técnicas radioterápicas focalizadas (radiocirugía, si la neoplasia responde a los requisitos mencionados anteriormente; téngase en cuenta que, por lo general, no se puede efectuar un segundo ciclo de radioterapia estándar a 60 Gy), o bien se le pueden administrar quimioterápicos diferentes de la temozolomida (a la que el paciente "ya no responde").[75]
Entre los quimioterápicos utilizados para la recurrencia pueden citarse la procarbazina, las nitrosoureas, el melfalán y el carboplatino. Los últimos estudios clínicos han mostrado una significativa actividad antitumoral con el uso de mitoxantrona[106] y la combinación de hidroxiurea con mesilato de imatinib.[107]
Otros ensayos clínicos sugieren el uso de inhibidores de los receptores del factor de crecimiento epidérmico,[108] el uso de agentes anti-angiogénicos,[109][110][111] o tratamientos combinados de radiofármacos inyectados localmente junto con quimioterápicos puros inyectados localmente.[112]
Todos estos estudios y protocolos están siendo evaluados por la comunidad científica. No obstante, un objetivo que se persigue es la identificación de un método práctico para caracterizar las clases de pacientes para las cuales un protocolo da mejores resultados, de modo que asignando al paciente a la clase más apropiada se le practique el protocolo de mayor eficacia, utilidad y mínimo impacto.
Pronóstico
La media de supervivencia desde el momento del diagnóstico, sin tratamiento, es de 3 meses, pero con él es común alcanzar una supervivencia de 1 o 2 años.[75] En la literatura, se habla de "supervivencia a largo plazo" a partir de los 3 años; un estudio muy citado sobre 279 pacientes que habían recibido un tratamiento completo agresivo, informó de que 5 de ellos (el 1,8 %) habían sobrevivido más de 3 años.[113] La edad avanzada (>60 años) constituye un factor de peor pronóstico. La muerte generalmente se debe a un edema cerebral o a la progresión de la hipertensión intracraneal.[114]
Una buena puntuación inicial en la escala de Karnofsky (KPS, por sus siglas en inglés), y la metilación de la MGMT están asociadas con una supervivencia más larga.[114] En glioblastomas, puede llevarse a cabo una prueba de ADN que determine si el promotor del gen MGMT está metilado o no. A los pacientes con un promotor MGMT metilado se les ha asociado un pronóstico a largo plazo significativamente más favorable respecto de los pacientes con un promotor MGMT no metilado,[115] pues los primeros, por ejemplo, pueden beneficiarse de una mejor respuesta al tratamiento con temozolomida.[116] Esta característica es intrínseca al ADN del paciente y en la actualidad no puede alterarse externamente.
También se han asociado mejores pronósticos a largo plazo a los pacientes sometidos a cirugía, radioterapia y quimioterapia con temozolomida. No obstante, aún queda mucho por saber acerca de por qué algunos pacientes de glioblastoma sobreviven durante más tiempo. A una mayor supervivencia con el glioblastoma multiforme están ligadas una edad de menos de 50 años, una resección de más del 98 %, el empleo de quimioterapia con temozolomida y una buena puntuación en la escala de Karnofsky.
Un estudio de 2003 dividió el pronóstico con ayuda del análisis de particiones recursivas (Recursive Partitioning Analysis, RPA) en tres subgrupos según la edad del paciente, el tipo de tratamiento y el índice de Karnofsky:[117]
| Clase RPA | Definición | Tiempo de supervivencia promedio | Tasa de supervivencia al año | Tasa de supervivencia a los 3 años | Tasa de supervivencia a los 5 años |
|---|---|---|---|---|---|
| III | Edad < 50, KPS ≥ 90 | 17,1 meses | 70 % | 20 % | 14 % |
| IV | Edad < 50, KPS < 90 | 11,2 meses | 46 % | 7 % | 4 % |
| Edad > 50, KPS ≥ 70, resección quirúrgica con buena función neurológica | |||||
| V + VI | Edad ≥ 50, KPS ≥ 70, resección quirúrgica con mala función neurológica | 7,5 meses | 28 % | 1 % | 0 % |
| Edad ≥ 50, KPS ≥ 70, sin resección quirúrgica | |||||
| Edad ≥ 50, KPS < 70 |
Referencias
- Tendencias actuales en el manejo del glioblastoma
- (en inglés) Kleihues P, Cavenee WK, eds. (2000), Pathology and genetics of tumours of the nervous system, World Health Organization classification of tumours. Lyon, France: IARC Press, ISBN 92-832-2409-4. En Internet puede accederse a la clasificación OMS de 2000 en la dirección «Copia archivada». Archivado desde el original el 11 de enero de 2014. Consultado el 31 de diciembre de 2010.. URL consultada el 31 de diciembre de 2010.
- Louis, David N.; Ohgaki, Hiroko; Wiestler, Otmar D.; Cavenee, Webster K.; Burger, Peter C.; Jouvet, Anne; Scheithauer, Bernd W.; Kleihues, Paul (12 de julio de 2007). «The 2007 WHO Classification of Tumours of the Central Nervous System». Acta Neuropathologica (en inglés) 114 (2): 97-109. ISSN 0001-6322. PMC 1929165. PMID 17618441. doi:10.1007/s00401-007-0243-4. Consultado el 17 de octubre de 2020.
- (en inglés) Kleihues P et al (2000). Glioblastoma. In Kleihues P, Cavenee WK, eds (2000). Pathology and genetics of tumours of the nervous system, World Health Organization classification of tumours. Lyon, France: IARC Press, ISBN 92-832-2409-4.
- A. Templeton, S. Hofer, M. Topfer, A. Sommacal, C. Fretz, T. Cerny, S. Gillessen (20 de marzo de 2008). «Extraneural Spread of Glioblastoma - Report of Two Cases». Onkologie 31 (4): 192-194.
- (en inglés) Lantos PL, VandenBerg SR, Kleihues P (1996). Tumours of the Nervous System. In: Greenfield's Neuropathology, Graham DI, Lantod PL (eds), 6th ed. Arnold: London. pp. 583-879
- Van Meir, E. G.; Hadjipanayis, C. G.; Norden, A. D.; Shu, H. K.; Wen, P. Y.; Olson, J. J. (2010). «Exciting New Advances in Neuro-Oncology: The Avenue to a Cure for Malignant Glioma». CA: A Cancer Journal for Clinicians (en inglés) 60 (3): 166-93. PMC 2888474. PMID 20445000. doi:10.3322/caac.20069.
- «ICD-0-3 SEER SITE/HISTOLOGY VALIDATION LIST». Archivado desde el original el 13 de junio de 2011. Consultado el 2 de febrero de 2011.
- Louis, David N.; Perry, Arie; Reifenberger, Guido; von Deimling, Andreas; Figarella-Branger, Dominique; Cavenee, Webster K.; Ohgaki, Hiroko; Wiestler, Otmar D. et al. (2016-06). «The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary». Acta Neuropathologica (en inglés) 131 (6): 803-820. ISSN 0001-6322. doi:10.1007/s00401-016-1545-1. Consultado el 17 de octubre de 2020.
- Glioblastoma. Vleeschouwer, Steven de,. Brisbane, Australia. ISBN 978-0-9944381-2-6. OCLC 1017991944.
- M. Stefani (1996). Lezioni di anatomia patologica. Piccin. ISBN 88-299-1184-4
- (en alemán) Virchow R (1863). Die Krankhaften Geschwulste. Hirschwald: Berlin.
- (en inglés) Mallory FB (1914). Principles of Pathologic Histology. Saunders: Philadelphia
- (en inglés) Globus JH, Strauss I (1925). Spongioblastoma multiforme. Arch. Neurol. Psychiatry 14:139-151
- (en inglés) Bailey P, Cushing H (1926). A Classification of Tumors of the Glioma Group on a Histogenetic Basis with a Correlation Study of Prognosis. Lippincott: Philadelphia
- (en inglés) Zülch KJ (1986). Brain Tumors. Their Biology and Pathology. 3rd ed, Springer Verlag: Berlin Heidelberg.
- (en inglés) Russell DS, Rubinstein LJ (1989). Pathology of Tumours of the Nervous System. 5th ed, Edward Arnold: London.
- (en inglés) Scherer HJ (1940). Cerebral astrocytomas and their derivatives. Am. J Cancer 40: 159-198.
- (en inglés) Kernohan JW, Mabon RF, Svien HJ, Adson AW (1949). A simplified classification of gliomas. Proc Staff Meet Mayo Clin 24:71-75
- El glioblastoma es el tumor más común que afecta al cerebro. El meningioma, más frecuente que el glioblastoma, es un tumor (normalmente de histología benigna), que tiene su origen en la aracnoides (membrana que recubre el cerebro y la médula espinal), y que comprime el cerebro, pero rara vez lo invade. Estrictamente hablando, no es un tumor cerebral. Igual ocurre con los tumores de las glándulas pituitaria y pineal, que no son estrictamente parte del cerebro. De hecho, la definición precisa de tales tumores sería de tumores intracraneales. (Véase a este propósito: DeAngelis LM, Gutin PH, Leibel SA, Posner JB (2002). Intracranial Tumors: Diagnosis and Treatment. Informa Health Care. ISBN 1-901865-37-1.)
- (en inglés) Dohrmann GJ, Farwell JR, Flannery JT (1976). Glioblastoma multiforme in children. J Neurosurg 44: 442-448.
- (en inglés) Buetow PC, Smirniotopoulos JG, Done S (1990). Congenital brain tumors: a review of 45 cases Archivado el 23 de julio de 2008 en Wayback Machine.. AJR AM J Roentgenol 155: 587-593.
- (en inglés) Lee DY, Kim YM, Yoo SJ, Cho BK, Chi JG, Kim IO, Wang KC (1999). Congenital glioblastoma diagnosed by fetal sonography. Childs Nerv Syst 15: 197-201.
- (en inglés) Sylvestre G, Sherer DM (1998). Prenatal sonographic findings associated with malignant astrocytoma following normal early third-trimester ultrasonography. Am J Perinatal 15: 581-584.
- (en inglés) Doren M, Tercanli S, Gullotta F, Holzgreve W (1997). Prenatal diagnosis of a highly undifferentiated brain tumour - a case report and review of the literature. Prenat Diagn 17: 967-971.
- (en inglés) Lee TT, Manzano GR (1997). Third ventricular glioblastoma multiforme: case report. Neurosurg Rev 20: 291-294.
- (en inglés) Henson G.W. (1999). Glioblastoma multiforme and anaplastic gliomas: A patient guide Archivado el 16 de febrero de 2012 en Wayback Machine.. URL consultada el 16 de julio 2008.
- (en inglés) Pollak L, Walach N, Gur R, Schiffer J. (1998). Meningiomas after radiotherapy for tinea capitis — still no history. Tumori 1998;84:65-8
- (en inglés) Walter AW, Hancock ML, Pui CH, Hudson MM, Ochs JS, Rivera GK, Pratt CB, Boyett JM, Kun LE (1998). Secondary brain tumors in children treated for acute lymphoblastic leukemia at St Jude Children's Research Hospital. J Clin Oncol 1998;16:3761-7
- (en inglés) Kaplan S, Novikov I, Modan B (1997) Nutritional factors in the etiology of brain tumors: potential role of nitrosamines, fat, and cholesterol. Am J Epidemiol 1997;146:832-41.
- (en inglés) Salvatore JR, Weitberg AB, Mehta S (1996). Nonionizing electromagnetic fields and cancer: a review. Oncology (Huntingt) 1996;10:563-74.
- (en inglés) Inskip PD, Mellemkjaer L, Gridley G, Olsen JH (1998). Incidence of intracranial tumors following hospitalization for head injuries (Denmark). Cancer Causes Control 1998;9:109-16.
- (en inglés) Inskip PD, Tarone RE, Hatch EE, Wilcosky TC, Shapiro WR, Selker RG, Fine HA, Black PM, Loeffler JS, Linet MS (2001). Cellular-telephone use and brain tumors. N Engl J Med 2001;344:79-86.
- (en inglés) DeAngelis L.M. (2001). Brain Tumors. N Engl J Med Vol. 344(2):114-123, January 11, 2001
- (en inglés) Rice JM, Wilbourn JD (2000). Tumors of the nervous system in carcinogenic hazard identification (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).. Toxicol Pathol. 2000 Jan-Feb;28(1):202-14.
- (en inglés) Cordier S, Monfort C, Filippini G, Preston-Martin S, Lubin F, Mueller BA, Holly EA, Peris-Bonet R, McCredie M, Choi W, Little J, Arslan A (2004). Parental exposure to polycyclic aromatic hydrocarbons and the risk of childhood brain tumors: The SEARCH International Childhood Brain Tumor Study. Am J Epidemiol. 2004 Jun 15;159(12):1109-16.
- (en italiano) G. Filippini (2006). Epidemiologia dei tumori cerebrali (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).. Mediterranean School of Oncology, Roma 6-7 de julio 2006. URL consultato il 10-11-2008.
- (en italiano) Macchi G. (1981 [I ed], 2005 [II ed]). Malattie del sistema nervoso. PICCIN Editore. ISBN 88-299-1739-7.
- (en italiano) Tomei G, Anzelmo V, Carbone M (2006). Agenti cancerogeni e sistema nervoso centrale (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).. 25 de marzo de 2006. XXI Giornata Romana di Medicina del Lavoro. Scuola di Specializzazione Medicina del Lavoro "La Sapienza".
- (en inglés) Nottebohm F (2004). The road we travelled: discovery, choreography, and significance of brain replaceable neurons. Ann. N. Y. Acad. Sci. 1016, 628–658.
- (en inglés) Reynolds BA & Weiss S (1992). Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255, 1707–1710.
- (en inglés) Gould E, McEwen BS, Tanapat P, Galea, LA & Fuchs E (1997). Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J. Neurosci. 17, 2492–2498.
- (en inglés) Gould E, Tanapat P, McEwen, BS, Flugge G & Fuchs E (1998). Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. Proc. Natl Acad. Sci. USA 95, 3168–3171.
- (en inglés) Eriksson PS, Perfilieva E, Björk-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH (1998). Neurogenesis in the adult human hippocampus. Nature Med. 4, 1313–1317.
- (en inglés) Lie DC, Song H, Colamarino SA, Ming GL & Gage FH (2004). Neurogenesis in the adult brain: new strategies for central nervous system diseases. Annu. Rev. Pharmacol. Toxicol. 44, 399–421.
- (en inglés) Vescovi AL, Galli R and Reynolds BA (2006). Brain tumour stem cells. Nature Reviews Cancer 6, 425-436 (Junio de 2006).
- (en inglés) Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA (2002). Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 39, 193–206.
- (en inglés) Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003). Identification of a cancer stem cell in human brain tumors. Cancer Res. 63, 5821–5828.
- (en inglés) Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A (2004). Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 64, 7011–7021.
- (en inglés) Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD y Rich JN (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Publicado el 18 de octubre de 2006.
- (en inglés) Rich JN (2007). Cancer Stem Cells in Radiation Resistance. Cancer Research 67, 8980-8984, 1 de octubre de 2007.
- (en inglés) Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL y Yu JS (2006). Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Molecular Cancer 2006, 5:67. Publicado el 2 de diciembre de 2006.
- (en inglés) Salmaggi A, Boiardi A, Gelati M, Russo A, Calatozzolo C, Ciusani E, Sciacca FL, Ottolina A, Parati EA, La Porta C, Alessandri G, Marras C, Croci D, De Rossi M (2006). Glioblastoma-derived tumorospheres identify a population of tumor stem-like cells with angiogenic potential and enhanced multidrug resistance phenotype. Glia. 2006 Dec;54(8):850-60. Epub 2006 Sep 15.
- (en inglés) Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC, Hainfellner JA, Heppner FL, Dietrich PY, Zimmer Y, Cairncross JG, Janzer RC, Domany E, Delorenzi M, Stupp R, Hegi ME (2008). Stem cell-related "self-renewal" signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008 Jun 20;26(18):3015-24.
- (en inglés) Hill RP (2006). Identifying Cancer Stem Cells in Solid Tumors: Case Not Proven. Cancer Research 66, 1891-1896, February 15, 2006.
- (en inglés) Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A (2007). Tumor growth need not be driven by rare cancer stem cells. Science. 2007 Jul 20;317(5836):337.
- (en inglés) Jin F, Zhao L, Zhao HY, Guo SG, Feng J, Jiang XB, Zhang SL, Wei YJ, Fu R, Zhao JS (2008). Paradoxical expression of anti-apoptotic and MRP genes on cancer stem-like cell isolated from TJ905 glioblastoma multiforme cell line. Cancer Invest. 2008 May;26(4):338-43.
- (en inglés) Yoo MH, Hatfield DL (2008). The Cancer Stem Cell Theory: Is It Correct? (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).. Mol Cells. 2008 Aug 14;26(5).
- (en inglés) Prestegarden L, Enger PØ (2010). Cancer stem cells in the central nervous system--a critical review. Cancer Res. 2010 Nov 1;70(21):8255-8. Epub 2010 Oct 19.
- (en inglés) Kleihues P, Burger PC, Aldape KD, Brat DJ, Biernat W, Bigner DD, Nakazato Y, Plate KH, Giangaspero F, von Deimling A, Ohgaki H, Cavenee WK (2007). Glioblastoma. (en inglés) Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2007). World Health Organization Classification of Tumours of the Central Nervous System. IARC, Lyon.
- (en inglés) Wilkening S, Bermejo JL, Burwinkel B, Klaes R, Bartram CR, Meindl A, Bugert P, Schmutzler RK, Wappenschmidt B, Untch M, Hemminki K, Försti A (2006). The Single Nucleotide Polymorphism IVS1+309 in 'Mouse Double Minute 2' Does Not Affect Risk of Familial Breast Cancer. Cancer Research 66, 646-648, January 15, 2006
- (en inglés) Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, Pandolfi PP, Jiang X (2007). NEDD4-1 Is a Proto-Oncogenic Ubiquitin Ligase for PTEN. Cell 128: 129-139.
- (en inglés) Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, Tempst P, Chi SG, Kim HJ, Misteli T, Jiang X, Pandolfi PP (2007). Ubiquitination Regulates PTEN Nuclear Import and Tumor Suppression. Cell 128: 141-156.
- (en inglés) Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y (2007). Essential Role for Nuclear PTEN in Maintaining Chromosomal Integrity. Cell 128: 157-170
- (en inglés) Kleihues P and Ohgaki H (1999). Primary and secondary glioblastomas: From concept to clinical diagnosis. Neuro-oncol 1999 January; 1(1): 44-51
- (en inglés) Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schüler D, Probst-Hensch NM, Maiorka PC, Baeza N, Pisani P, Yonekawa Y, Yasargil MG, Lütolf UM, Kleihues P (2004). Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004 Oct 1;64(19):6892-9
- Ohgaki H, Kleihues P (2007). Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007 May;170(5):1445-53
- Para un tratamiento completo y actualizado, puede consultarse por Internet (en inglés) «Copia archivada». Archivado desde el original el 23 de junio de 2008. Consultado el 6 de agosto de 2008.. URL consultada el 24 de agosto de 2008.
- Burger PC, Kleihues P (1989). Cytologic composition of the untreated glioblastoma with implications for evaluation of needle biopsies. Cancer 63: 2014-2023
- Fujisawa H, Kurrer M, Reis RM, Yonekawa Y, Kleihues P, Ohgaki H (1999). Acquisition of the Glioblastoma Phenotype during Astrocytoma Progression Is Associated with Loss of Heterozygosity on 10q25-qter. American Journal of Pathology. 1999;155:387-394.
- (en italiano) Bruzzone MG, Farina L. Imaging dei gliomi cerebrali. URL consultada el 22-09-2008.
- (en italiano) Biagini C, Gavelli G (1999). Radiobiologia e radioprotezione. PICCIN Editore. ISBN 88-299-1463-0.
- (en inglés) Hustinx, R., Pourdehnad, M., Kaschten, B., & Alavi, A. (2005). PET imaging for differentiating recurrent brain tumor from radiation necrosis. Radiologic Clinics, 43(1), 35-37.
- Smirniotopoulos, J. G.; Murphy, F. M.; Rushing, E. J.; Rees, J. H.; Schroeder, J. W. (2007). «From the Archives of the AFIP: Patterns of Contrast Enhancement in the Brain and Meninges». Radiographics 27 (2): 525-51. PMID 17374867. doi:10.1148/rg.272065155.
- (en inglés) DeAngelis LM, Loeffler JS, Adam N. Mamelak AN (2007). Primary and Metastatic Brain Tumors. En Pazdur R, Coia LR, Hoskins WJ, and Wagman LD (2007). Cancer Management: A Multidisciplinary Approach, 10th Edition. URL consultada el 24-09-2008.
- Lawson, H. Christopher; Sampath, Prakash; Bohan, Eileen; Park, Michael C.; Hussain, Namath; Olivi, Alessandro; Weingart, Jon; Kleinberg, Lawrence et al. (2006). «Interstitial chemotherapy for malignant gliomas: the Johns Hopkins experience». Journal of Neuro-Oncology 83 (1): 61-70. PMID 17171441. doi:10.1007/s11060-006-9303-1.
- «Brain Tumours & Cancer | Signs, Symptoms, Types and Treatment». patient.info (en inglés). Consultado el 14 de noviembre de 2021.
- (en inglés) Chang SM, Parney IF, Huang W, Anderson FA Jr, Asher AL, Bernstein M, Lillehei KO, Brem H, Berger MS, Laws ER; Glioma Outcomes Project Investigators (2005). Patterns of care for adults with newly diagnosed malignant glioma. JAMA 2005 Feb 2;293(5):557-64.
- (en inglés) Williams BA (2008). Treatment Options for Glioblastoma and other Gliomas (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).. BrainLife Newsletter Volume 7, Number 19 - 1 October 2008.
- (en inglés) Barnholtz-Sloan JS, Williams VL, Maldonado JL, Shahani D, Stockwell HG, Chamberlain M, Sloan AE (2008). Patterns of care and outcomes among elderly individuals with primary malignant astrocytoma J Neurosurg. 2008 Apr;108(4):642-648.
- (en inglés) Forsyth PA, Weaver S, Fulton D, Brasher PMA, Sutherland G, Stewart D, Hagen NA (2003). Prophylactic Anticonvulsants in Patients with Brain Tumour. The Canadian Journal of Neurological Sciences, Volume 30, Number 2 / May 2003.
- (en inglés) Laws ER, Parney IF, Huang W, Anderson F, Morris AM, Asher A, Lillehei KO, Bernstein M, Brem H, Sloan A, Berger MS, Chang S; Glioma Outcomes Investigators (2003). Survival following surgery and prognostic factors for recently diagnosed malignant glioma: data from the Glioma Outcomes Project (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).. J Neurosurg. 2003 Sep;99(3):467-73.
- R, Díez Valle (2019 Feb). «Established and Emerging Uses of 5-ALA in the Brain: An Overview». Journal of neuro-oncology (en inglés). Consultado el 16 de abril de 2020.
- (en inglés) Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ, Lin JW, Chute DJ, Mischel PS, Cloughesy TF, Roth MD (2005). Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005 Aug 1;11(15):5515-25.
- (en inglés) Tatter SB, Shaw EG, Rosenblum ML, Karvelis KC, Kleinberg L, Weingart J, Olson JJ, Crocker IR, Brem S, Pearlman JL, Fisher JD, Carson K, Grossman SA; New Approaches to Brain Tumor Therapy Central Nervous System Consortium (2003). An inflatable balloon catheter and liquid 125I radiation source (GliaSite Radiation Therapy System) for treatment of recurrent malignant glioma: multicenter safety and feasibility trial (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).. J Neurosurg. 2003 Aug;99(2):297-303.
- (en inglés) Chiocca EA, Broaddus WC, Gillies GT, Visted T, Lamfers ML (2004). Neurosurgical delivery of chemotherapeutics, targeted toxins, genetic and viral therapies in neuro-oncology. J Neurooncol. 2004 Aug-Sep;69(1-3):101-17.
- (en inglés) Perry J, Chambers A, Spithoff K, Laperriere N (2007). Gliadel wafers in the treatment of malignant glioma: a systematic review. Curr Oncol. 2007 Oct;14(5):189-94.
- (en inglés) Attenello FJ, Mukherjee D, Datoo G, McGirt MJ, Bohan E, Weingart JD, Olivi A, Quinones-Hinojosa A, Brem H (2008). Use of Gliadel (BCNU) Wafer in the Surgical Treatment of Malignant Glioma: A 10-Year Institutional Experience. Ann Surg Oncol. 2008 Jul 18.
- (en inglés) Walker MD, Strike TA, Sheline GE (1979). An analysis of dose-effect relationship in the radiotherapy of malignant gliomas. Int J Radiat Oncol Biol Phys. 1979 Oct;5(10):1725-31. [Median OS: no RT 18 weeks vs. 50 Gy 28 weeks vs. 55 Gy 36 weeks vs. 60 Gy 42 weeks]
- (en inglés) Cook B, Dvorak T et al. Radiation Oncology. Wikibook. Section High Grade Gliomas Adjuvant Therapy.
- (en inglés) Buatti J, Ryken TC, Smith MC, Sneed P, Suh JH, Mehta M, Olson JJ (2008). Radiation therapy of pathologically confirmed newly diagnosed glioblastoma in adults. J Neurooncol. 2008 Sep;89(3):313-37. Epub 2008 Aug 20.
- (en inglés) Roa W, Brasher PM, Bauman G, Anthes M, Bruera E, Chan A, Fisher B, Fulton D, Gulavita S, Hao C, Husain S, Murtha A, Petruk K, Stewart D, Tai P, Urtasun R, Cairncross JG, Forsyth P (2004). Abbreviated course of radiation therapy in older patients with glioblastoma multiforme: a prospective randomized clinical trial. J Clin Oncol. 2004 May 1;22(9):1583-8. Epub 2004 Mar 29.
- (en inglés) Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. (2005). Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. New England Journal of Medicine 352(10), 987-996.
- (en inglés) Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005 Mar 10;352(10):997-1003.
- (en inglés) Hegi ME, Liu L, Herman JG, Stupp R, Wick W, Weller M, Mehta MP, Gilbert MR (2008). Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol. 2008 Sep 1;26(25):4189-99.
- (en inglés) Brandes AA, Franceschi E, Tosoni A, Blatt V, Pession A, Tallini G, Bertorelle R, Bartolini S, Calbucci F, Andreoli A, Frezza G, Leonardi M, Spagnolli F, Ermani M (2008). MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J Clin Oncol. 2008 May 1;26(13):2192-7.
- (en inglés) Hall W, Christie M, Currow D (2005). Cannabinoids and cancer: causation, remediation, and palliation. Lancet Oncol 6: 35–42 (January 2005).
- (en inglés) Guzmán M (2003). Cannabinoids: potential anticancer agents. Nat Rev Cancer 3: 745–755 (October 2003).
- (en inglés) Massi P, Vaccani A, Ceruti S, Colombo A, Abbracchio MP, Parolaro D (2004). Antitumor Effects of Cannabidiol, a Nonpsychoactive Cannabinoid, on Human Glioma Cell Lines. J Pharmacol Exp Ther. 2004 Mar;308(3):838-45. Epub 2003 Nov 14.
- (en inglés) Blázquez C, Casanova ML, Planas A, Gómez Del Pulgar T, Villanueva C, Fernández-Aceñero MJ, Aragonés J, Huffman JW, Jorcano JL, Guzmán M (2003). Inhibition of tumor angiogenesis by cannabinoids. FASEB J. 2003 Mar;17(3):529-31. Epub 2003 Jan 2.
- (en inglés) Guzmán M, Duarte MJ, Blázquez C, Ravina J, Rosa MC, Galve-Roperh I, Sánchez C, Velasco G, González-Feria L (2006). A pilot clinical study of Delta9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br J Cancer. 2006 Jul 17;95(2):197-203. Epub 2006 Jun 27.
- (en inglés) Aguado T, Carracedo A, Julien B, Velasco G, Milman G, Mechoulam R, Alvarez L, Guzmán M, Galve-Roperh I (2007). Cannabinoids Induce Glioma Stem-like Cell Differentiation and Inhibit Gliomagenesis Archivado el 18 de junio de 2008 en Wayback Machine.. J Biol Chem. 2007 Mar 2;282(9):6854-62. Epub 2007 Jan 2.
- http://neuro-oncology.oxfordjournals.org/content/14/suppl_6/vi43.abstract
- http://www.cun.es/profesionales/adenovirus-oncoliticos-estrategia-terapeutica-superar-la-resistencia-quimioterapia-exhiben-celulas-m
- (en inglés) Hou LC, Veeravagu A, Hsu AR, Tse VC. (2006). Recurrent Glioblastoma Multiforme: A Review of Natural History and Management Options. Neurosurg Focus. 2006;20(4):E5.
- (en inglés) Boiardi A, Silvani A, Eoli M, Lamperti E, Salmaggi A, Gaviani P, Fiumani A, Botturi A, Falcone C, Solari A, Filippini G, Di Meco F, Broggi G (2008). Treatment of recurrent glioblastoma: can local delivery of mitoxantrone improve survival?. J Neurooncol. 2008 Feb 19.
- (en inglés) Reardon DA, Egorin MJ, Quinn JA, Rich JN, Gururangan S, Vredenburgh JJ, Desjardins A, Sathornsumetee S, Provenzale JM, Herndon JE 2nd, Dowell JM, Badruddoja MA, McLendon RE, Lagattuta TF, Kicielinski KP, Dresemann G, Sampson JH, Friedman AH, Salvado AJ, Friedman HS (2005). Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol. 2005 Dec 20;23(36):9359-68.
- (en inglés) Combs SE, Heeger S, Haselmann R, Edler L, Debus J and Schulz-Ertner D (2006). Treatment of primary glioblastoma multiforme with cetuximab, radiotherapy and temozolomide (GERT) - phase I/II trial: study protocol. BMC Cancer 2006, 6:133; Published: 18 May 2006.
- (en inglés) Reardon DA, Desjardins A, Rich JN, Vredenburgh JJ (2008). The emerging role of anti-angiogenic therapy for malignant glioma. Curr Treat Options Oncol. 2008 Feb;9(1):1-22. Epub 2008 Feb 7.
- (en inglés) Marx GM, Pavlakis N, McCowatt S, Boyle FM, Levi JA, Bell DR, Cook R, Biggs M, Little N, Wheeler HR (2001). Phase II study of thalidomide in the treatment of recurrent glioblastoma multiforme. J Neurooncol 2001 Aug;54(1):31-8.
- (en inglés) Baumann F, Bjeljac M, Kollias SS, Baumert BG, Brandner S, Rousson V, Yonekawa Y, Bernays RL (2004). Combined Thalidomide and Temozolomide Treatment in Patients with Glioblastoma Multiforme. Journal of Neuro-Oncology, 67 (1-2): 191-200, March, 2004 - April, 2004.
- (en inglés) Boiardi A, Bartolomei M, Silvani A, Eoli M, Salmaggi A, Lamperti E, Milanesi I, Botturi A, Rocca P, Bodei L, Broggi G and Paganelli G (2005). Intratumoral delivery of mitoxantrone in association with 90-Y radioimmunotherapy (RIT) in recurrent glioblastoma. Journal of Neuro-Oncology, Vol 72, Number 2, April, 2005, Pages 125-131, Online Date May 31, 2005.
- (en inglés) Scott JN, Rewcastle NB, Brasher PM, Fulton D, Hagen NA, MacKinnon JA, Sutherland G, Cairncross JG, Forsyth P (1998). Long-term glioblastoma multiforme survivors: a population-based study. Can J Neurol Sci. 1998 Aug;25(3):197-201.
- Krex, D.; Klink, B.; Hartmann, C.; Von Deimling, A.; Pietsch, T.; Simon, M.; Sabel, M.; Steinbach, J. P. et al. (2007). «Long-term survival with glioblastoma multiforme». Brain (en inglés) 130 (Pt 10): 2596-606. PMID 17785346. doi:10.1093/brain/awm204.
- Martinez, Ramon; Schackert, Gabriele; Yaya-Tur, Ricard; Rojas-Marcos, Iñigo; Herman, James G.; Esteller, Manel (2006). «Frequent hypermethylation of the DNA repair gene MGMT in long-term survivors of glioblastoma multiforme». Journal of Neuro-Oncology (en inglés) 83 (1): 91-3. PMID 17164975. doi:10.1007/s11060-006-9292-0.
- Franco-Hernández C, Martínez-González V, Rey JA. Biología molecular de los glioblastomas Archivado el 4 de marzo de 2016 en Wayback Machine.. Neurocirugía 2007; 18: 373-382.
- Shaw, E; Seiferheld, W; Scott, C; Coughlin, C; Leibel, S; Curran, W; Mehta, M (2003). «Reexamining the radiation therapy oncology group (RTOG) recursive partitioning analysis (RPA) for glioblastoma multiforme (GBM) patients». International Journal of Radiation OncologyBiologyPhysics (en inglés) 57 (2): S135-6. doi:10.1016/S0360-3016(03)00843-5.
Bibliografía
- Escalona-Zapata, Julio; Bello, M. J. (1996). Tumores del sistema nervioso central. Editorial Complutense. ISBN 8489365598.
- Reyes Oliveros, Francisco (2007). Gliomas del encéfalo. Univ. Santiago de Compostela. ISBN 8497508130.
- Aguirre Cruz, María Lucinda; Sotelo Morales, Julio (2008). Tumores cerebrales. Volumen 1. Ed. Médica Panamericana. ISBN 9687988843.
- Ray, Swapan K. (2009). Glioblastoma: Molecular Mechanisms of Pathogenesis and Current Therapeutic Strategies (en inglés). Springer. ISBN 1441904093.
- Markert, James (2005). Glioblastoma Multiforme (en inglés). Jones & Bartlett Learning. ISBN 0763726400.
- Tonn, Jörg-Christian; Westphal, Manfred; Rutka, James T. (2009). Oncology of CNS Tumors (en inglés). Springer. ISBN 364202873X.
Enlaces externos
 Wikimedia Commons alberga una galería multimedia sobre Glioblastoma.
Wikimedia Commons alberga una galería multimedia sobre Glioblastoma.- Diagnóstico y tratamiento del glioblastoma multiforme - Clínica Neuros
- Tumor cerebral primario en adultos - MedlinePlus
- Glioblastoma. Síntomas, causas y tratamiento
- Glioblastoma multiforme - Dr. J. Sales Llopis
- GBM and Anaplastic Astrocytomas - American Brain Tumor Association (en inglés)
- Banco de imágenes (en inglés)
| Control de autoridades |
|
|---|
 Datos: Q282142
Datos: Q282142- Multimedia: Glioblastoma / Q282142