Globósido
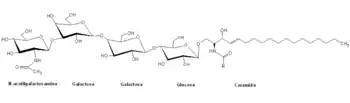
Los Globósidos son oligosacáridos de ceramida que contienen dos o más residuos de azúcar, generalmente: galactosa, glucosa y N-acetilgalactosamina. Los oligosacáridos de ceramida son compuestos neutros ya que no tienen carga a PH 7 y además no contienen grupos amino libres.
Características
Los globósidos son una clase de lípidos complejos que pertenecen a la familia de los glucoesfingolípidos (glucolípidos). Se conocen cuatro tipos de glucoesfingolípidos: los cerebrósidos, importantes componentes del músculo y del sistema nervioso central y periférico que forman parte de la vaina de mielina de los nervios, los gangliósidos, lípidos complejos ricos en hidratos de carbono con una o más unidades de N-acetilneuramínico o ácido siálico (NANA) que se encuentran por lo general en la superficie externa de las membranas celulares del tejido nervioso, los sulfátidos, ésteres de ácido sulfúrico de los galactocerebrósidos y finalmente los globósidos.
Biosíntesis
Una serie de glucosiltransferasas cataliza la biosíntesis de los globósidos. La síntesis comienza con la transferencia de una unidad galactosilo a partir de UDP-Galactosa a un glucocerebrósido para formar un enlace β (1-4) Dado que este enlace es el mismo que une la glucosa y la galactosa en la lactosa, este glucolípido suele designarse como una lactosil ceramida. Esta última es el precursor de los globósidos y de los gangliósidos.
A continuación se adiciona otra unidad galactosilo a partir de UDP-Galactosa, pero en este caso el residuo galactosa terminal adopta la configuración anomérica α y da lugar a la trihexosil ceramida. Para terminar de formar un globósido, una unidad N-acetilgalactosilo se une de forma secuencial a la trihexosil ceramida a partir de UDP-N-acetilgalactosilo.

Función
Los globósidos están implicados en diversas funciones de reconocimiento en la superficie de la membrana. Entre ellas, son el antígeno P del eritrocito, que es el principal receptor del parvovirus B-19 humano. Estudios recientes sugieren que los globósidos, aparte de ser el receptor primario de B-19 actúan también como mediador de los cambios de la cápsula vírica requeridos para la internalización del virus.[1] Hay algunos pocos individuos que no expresan el globósido en su membrana, por lo que son resistentes a la infección por B-19.[2]
Localización
Los globósidos forman parte de la bicapa lipídica de la membrana celular, con su parte glucídica orientada hacia el exterior de la membrana plasmática. Los globósidos sólo se encuentran en la membrana de las células progenitoras de los eritrocitos, en los miocitos del corazón, en las células endoteliales y en células de algunos órganos como el hígado, riñón y bazo. Cabe destacar que la membrana de las mitocondrias y del retículo endoplasmático no presenta globósidos ni ningún otro tipo de glucolípidos.
Enfermedades asociadas
Existen algunas enfermedades degenerativas asociadas a un desorden en el metabolismo de los globósidos debido a una falta o deficiencia de las enzimas encargadas de su degradación en los lisosomas.[3] Todas ellas se incluyen dentro de las lipidosis o enfermedades causadas por almacenamiento de lípidos. Entre ellas cabe destacar:
Enfermedad de Sandhoff
La enfermedad de Sandhoff es una enfermedad hereditaria autosómica recesiva, ocasionada por la deficiencia de las isoenzimas β-hexosaminidasa A y B debido a una mutación en el gen HEXB, localizado en el brazo largo del cromosoma 5. Estas enzimas, en su estado normal, son las encargadas de romper el enlace β que une la N-acetilgalactosamina, degradando así los globósidos a trihexosilceramida. Consecuentemente, un defecto en estas enzimas provoca la acumulación a niveles tóxicos de globósidos; particularmente en las neuronas del cerebro y de la médula espinal, causando su progresiva deterioración y destrucción.
Las manifestaciones clínicas recuerdan a la enfermedad de Tay-Sachs[4] pero con una progresión mucho más rápida. La forma infantil se presenta en los 4 primeros meses de vida y se caracteriza por retraso psicomotor, pérdida de visión y audición, hipotonía seguida por espasticidad, mioclono (contracciones de un músculo parecidas a una descarga), convulsiones, macrocefalia (cabeza anormalmente agrandada), y manchas rojo-cereza en los ojos. Otros síntomas pueden incluir infecciones respiratorias, soplos cardíacos, características faciales de muñeca y agrandamiento del hígado y el bazo. Los bebés afectados con esta enfermedad suelen fallecer a temprana edad ya que actualmente no existe ningún tratamiento para evitar el progreso de ésta enfermedad.
Enfermedad de Fabry
La enfermedad de Fabry es una enfermedad hereditaria recesiva ligada al cromosoma X derivada de mutaciones en el gen GLA que codifica la enzima α-galactosidasa. Sin cantidades adecuadas de α-galactosidasa no se puede degradar la trihexosilceramida, derivada de los globósidos, en partículas más pequeñas (lactosil ceramida), acumulándose así en vasos sanguíneos, tejidos del hígado, riñón, cerebro y corazón, lo que afecta su correcto funcionamiento.
Entre los síntomas asociados a esta enfermedad se pueden encontrar: dolor en las extremidades, anhidrosis (disminución en la sudoración), intolerancia al calor y al frío, aparición de angioqueratomas (grupos de puntos de color rojo granate), opacidad de la córnea así como otros problemas digestivos, cardíacos, cerebrales, renales y nerviosos que son lo que acaban causando la muerte.
Referencias
- Bönsch C, Zuercher C, Lieby P, Kempf C, Ros C (2010). «The globoside receptor triggers structural changes in the B19 virus capsid that facilitate virus internalization». Journal of Virology 84 (22). PMID 20826697. doi:10.1128/JVI.01143-10.
- Hellberg A, Poole J, Olsson ML. (2002). «Molecular basis of the globoside-deficient P(k) blood group phenotype. Identification of four inactivating mutations in the UDP-N-acetylgalactosamine: globotriaosylceramide 3-beta-N-acetylgalactosaminyltransferase gene.». The Journal of biological chemistry 277 (22). PMID 12023287. doi:10.1074/jbc.M203047200.
- http://www.glycoactive.com/GSL.pdf Archivado el 5 de marzo de 2016 en Wayback Machine.] Los glicoesfingolípidos y defectos lisosomales
- Sandhoff K, Andreae U, Jatzkewitz H (1968). «Deficient hexozaminidase activity in an exceptional case of Tay-Sachs disease with additional storage of kidney globoside in visceral organs». Life Sci. 7 (6): 283-288. PMID 5651108. doi:10.1016/0024-3205(68)90024-6.
Enlaces externos
 Wikimedia Commons alberga una galería multimedia sobre Globósido.
Wikimedia Commons alberga una galería multimedia sobre Globósido.- MeSH: Globosides (en inglés) (en inglés)
- The National Academy of Sciences of the USA (en inglés)
- Bioquímica médica (en español)
Enfermedades asociadas:
- What is Sandhoff disease? (en inglés)
- Genetics Home Reference: Sandhoff disease (en inglés)
- GeneReview entry on Fabry disease by NIH (en inglés)
- Fabry disease at NLM Genetics Home Reference (en inglés)