Oncogén
Un oncogén es un gen anormal o activado que procede de la mutación de un alelo de un gen normal llamado protooncogén.[1] Los oncogenes son los responsables de la transformación de una célula normal en una maligna que desarrollará un determinado tipo de cáncer. En el ser humano se han identificado y secuenciado más de 62 oncogenes en los diferentes cromosomas del genoma, formando un conjunto muy heterogéneo de genes.
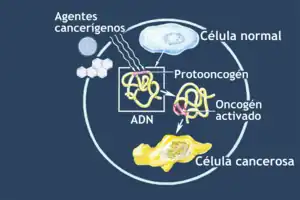
En un individuo humano sano, existen más de 30000 billones de células que viven en un condominio interdependiente, regulando de forma mutua su proliferación, para asegurar que el tamaño de los diferentes órganos está coordinado y de acuerdo con la talla del individuo. Por ello, las células solo proliferan cuando reciben señales muy específicas que provienen de otras células vecinas. Las células cancerosas, sin embargo, violan este esquema: ignoran todas las señales que reciben del exterior, y siguen sus propios esquemas de proliferación, invadiendo no solo los espacios adyacentes, sino también sitios alejados del lugar de origen, a través del proceso de metástasis.[2] Desde este punto de vista, las células cancerosas pueden considerarse como células "asociales", que no siguen las pautas del conjunto del organismo e incluso amenazan su supervivencia.
Todas las células de un tumor proceden de una única célula (pertenecen a un mismo clon), un ancestro común que en un momento dado (puede que décadas antes de la detección del tumor) inició un programa inadecuado de proliferación. Esta transformación maligna se produce por acumulación de mutaciones en un conjunto de genes muy específico. Existen dos clases de genes, que en conjunto representan una proporción muy pequeña del conjunto del genoma, que juegan un papel fundamental en el inicio de la progresión tumoral. En condiciones normales, estos genes participan en la regulación del ciclo vital de la célula: el conjunto de sucesos que definen cuándo una célula debe crecer y proliferar. Los genes reguladores pueden realizar dos tipos de funciones:
- activar los procesos dirigidos hacia el crecimiento y la proliferación -- estos genes se denominan protooncogenes; contribuyen a la progresión tumoral cuando sufren mutaciones que los activan de forma permanente o constitutiva, es decir, cuando se produce una ganancia de función; este tipo de mutación tiene un efecto dominante: basta que uno de los dos alelos de la célula esté mutado para que aparezca la actividad;
- inhibir dichos procesos -- son los denominados genes supresores de tumores; en este caso, intervienen en el proceso tumoral si sufren mutaciones que los inactivan, es decir, si se produce una pérdida de función; este tipo de mutación tiene un efecto recesivo: para eliminar la actividad, tienen que estar mutados los dos alelos (véase también Hipótesis de Knudson).
Para que un cáncer pueda progresar y desarrollarse, deben producirse al menos media docena de mutaciones que afecten a varios genes reguladores.[2] Sin embargo, otros tipos de genes también pueden participar en la malignidad, facilitando la capacidad invasiva del tumor (por ejemplo, mutaciones en las proteínas del citoesqueleto que favorecen la motilidad celular).
Concepto de oncogén
Los oncogenes proceden de genes reguladores, los protooncogenes. Muchos protooncogenes participan en cascadas de señalización que reciben, integran y transmiten señales de proliferación provenientes del exterior, ejecutando programas específicos mediante la expresión de genes concretos que ponen en marcha la maquinaria celular de crecimiento y entrada en el ciclo celular. Esta señalización se transmite de una célula a otra en un tejido, y normalmente se inicia debido a la secreción de factores de crecimiento a partir de diferentes tipos celulares (por ejemplo, los fibroblastos durante la cicatrización). Estos factores de crecimiento pasan a través de los espacios intercelulares, y son reconocidos por receptores de membrana específicos para esa molécula. Los receptores de membrana son proteínas que presentan un extremo hacia el exterior celular y otro hacia el interior. Cuando un factor de crecimiento se asocia a su receptor, este transmite una señal hacia el citoplasma, produciendo un cambio en la conformación de una o varias proteínas, que se transmite en forma de cascada, hasta activar en el núcleo la expresión de los genes adecuados para responder a la señal emitida. Cuando se producen mutaciones que desregulan algunos de estos procesos, de manera que se mantienen activados cuando deberían permanecer detenidos, el crecimiento celular deviene anárquico.
Los protooncogenes son por tanto genes normales responsables de la codificación de proteínas nucleares, citoplasmáticas y de membrana, que intervienen en la homeostasis celular, es decir, en el mantenimiento del equilibrio de las funciones celulares, por lo que su nivel de expresión está estrictamente regulado. Muchos protooncogenes están muy expresados durante ciertas etapas del ciclo celular y/o muy relacionadas con determinadas fases del desarrollo embrionario.
En todas las células del organismo existen muchos protooncogenes y cuando un grupo de ellos se altera, puede precipitarse la transformación maligna de la célula o el desarrollo de un cáncer. Los protooncogenes existen en muchas especies de organismos pluricelulares, estando bien conservados entre diferentes especies, mientras que distintos protooncogenes pueden no ser parecidos dentro de una especie en concreto.
En algunos casos, los oncogenes virales proceden de genes celulares que en algún momento fueron secuestrados por el virus, y mutaron, dando como resultado un oncogén. Por lo tanto, a los oncogenes no mutados que se encuentran en las células normales, se les llama protooncogenes, y a los mutados, oncogenes. Los oncogenes se designan con tres letras, por ejemplo src por el virus del sarcoma de Rous. A la forma viral o maligna del oncogén se le antepone una v (v-src) y a la forma benigna, normal o celular se le antepone una c (c-src). Un gran número de oncogenes identificados en retrovirus entra dentro de este grupo, por ejemplo los oncogenes abl, erb-B, fes, fms, fos/jun, kit, raf, myc, H-ras, K-ras, rel y sis, además de src.
El descubrimiento y conocimiento de los oncogenes confirma que el cáncer es una enfermedad genética con las siguientes salvedades:
- El desarrollo del cáncer no es debido a la expresión de un solo oncogén. Es preciso la acumulación de varios oncogenes en una sola célula (teoría clonal) o un número determinado de oncogenes iguales en varias células para que se manifieste el cáncer.
- Los oncogenes no son la única causa del cáncer. El sistema inmune también es uno de los factores reguladores al eliminar células cancerosas (que manifiestan oncogenes) o por el contrario no reconocer a las células malignas y permitir su supervivencia y proliferación. El cáncer es un conjunto de enfermedades multifactoriales, por lo cual, los oncogenes no son la única causa.
Clasificación de los oncogenes
Los oncogenes pueden codificar proteínas que actúan a diferentes niveles de la cascada de señalización que activa la proliferación celular:[2]
Extracelular: exceso de producción de factores de crecimiento
En este caso, los oncogenes fuerzan a la célula a producir un exceso de factores de crecimiento; estos factores influyen no solo sobre las células vecinas, sino que además pueden activar la proliferación de las células que los produjeron:
Membrana: receptores modificados
Se producen versiones oncogénicas de receptores celulares para factores de crecimiento, que transmiten una señal de proliferación hacia el interior celular en ausencia de factores de crecimiento en el exterior:
- las células tumorales de mama a menudo expresan receptores Erb-B2 que funcionan de este modo;
- otros ejemplos son los oncogenes src o fms.
Citoplasma: cascadas de señalización constitutivas
Se generan versiones oncogénicas de proteínas citoplásmicas de la cascada de señalización que se mantienen activas siempre:
- el caso mejor estudiado es el de la familia de proteínas Ras; los productos de la familia Ras unen GTP, se asocian a GTPasas y actúan como transductores de señales para receptores de factores de crecimiento en la superficie celular; el oncogén Ras mutado actúa constitutivamente, uniendo siempre GTP; formas de Ras hiperactivo se encuentran presentes en un cuarto de todos los tumores humanos, incluyendo carcinomas (tumores epiteliales) de colon, páncreas y pulmón;
- proteínas citoplasmáticas con actividad kinasa:
- por ejemplo la proteína c-Raf puede ir al núcleo para ejercer la función recibida en la membrana activada, actuando como segundo mensajero; la forma oncogénica de Raf ha perdido las secuencias reguladoras del extremo amino y está constitutivamente activa;
- otro tipo, c-Crk, es una proteína citoplasmática que estabiliza las tirosina kinasas;
Núcleo: factores de transcripción o secuencias asociadas constitutivas
Se producen versiones oncogénicas de factores de transcripción o secuencias asociadas que funcionan en todo momento:
- la alteración oncogénica de los factores de transcripción los convierte en proteínas oncogénicas con pérdida de sus elementos negativos o pérdida de su dominio activo (mutación dominante negativa):
- es el caso de la familia de factores de transcripción myc); normalmente, las células solo producen Myc cuando son estimuladas mediante factores de crecimiento, y una vez producidos estimulan la transcripción de genes que activan la proliferación celular; sin embargo, en muchos tipos de cáncer (sobre todo en los asociados con los tejidos hematopoyéticos), los niveles de Myc permanecen elevados aún en ausencia de factores de crecimiento;
- otros oncogenes que codifican para factores de transcripción constitutivos son myb, fos, jun, erb-A y rel.
- modificación de secuencias reguladoras que están próximas a genes codificantes, compuestos por segmentos cortos de ADN que sirven como diana para los factores de transcripción que activan los genes codificantes; muchas de estas secuencias reguladoras se localizan fuera de las secuencias codificadoras de proteínas, en la zona del ADN no codificante o ADN basura, que puede representar el 97% del genoma humano.
Aunque los genes nucleares son capaces de perpetuar la proliferación celular, no tienen capacidad de formar tumores malignos. Para adquirir la capacidad tumorogénica es preciso la activación de un segundo oncogén, generalmente citoplasmático, por lo que para que aparezca un tumor maligno es necesario la activación de varios oncogenes.
Según la función de la proteína codificada
- Oncogenes que codifican proteínas G: el más común es el Ras. Este gen codifica una proteína G monomérica que, en el protooncogen, al estar desactivada, hidroliza el GTP a GDP, desactivando la proliferación celular. El oncogen, en cambio, la mantiene en su forma activada. Así, no puede hidrolizar el GTP y la proliferación celular continúa.
- Oncogenes que codifican factores de crecimiento o sus receptores: el oncogen sis (virus del sarcoma de simio) codifica el factor de crecimiento PDGF, cuya producción excesiva estimula la proliferación celular. El erb-B (eritroblastosis aviar) rige la formación de un receptor para el factor de crecimiento EGF, que al estar alterado actúa como si estuviera permanentemente unido a EGF, estimulando la proliferación celular.
- Oncogenes que codifican proteínas quinasas de serina-treonina y de tirosina: Raf es una quinasa de serina-treonina que actúa en el inicio de la cascada del AMP cíclico, que es la vía primaria del control de la proliferación celular. El oncogen mantiene a Raf en la forma activa, evitando que se desactive la proliferación. El gen Src es una quinasa de tirosina que produce señales intracelulares, muchas de ellas relacionadas con la proliferación celular.
- Oncogenes que codifican factores de transcripción nuclear: el gen Myc causa el paso de G0 a G1 en una proliferación celular que no debería ocurrir.
- Oncogenes que codifican productos que afectan a la apoptosis: el gen Bcl-2, al estar sobreexpresado, suprime la apoptosis.
Activación de los oncogenes
La activación de un protooncogén y su transformación a un oncogén se produce por mutaciones ocasionadas por causas físicas como las radicaciones ionizantes, causas químicas como los carcinógenos, causas biológicas como los virus oncogénicos o causas hereditarias, por mutaciones transmitidas a lo largo de generaciones o por fallo en alguno de los mecanismos de reparación del ADN.
Los mecanismos por los que un protooncogén puede ser transformado en un oncogén son cuantitativos y cualitativos.
Mecanismos cuantitativos
- Inserción de un promotor viral: Algunos retrovirus contienen una secuencia promotora llamada LTR (Long Terminal Repeat, en inglés), que cuando es incorporado al ADN de la célula infectada adyacente a las secuencias reguladoras de un protooncogén, se produce un aumento en la expresión de ese gen que queda bajo el control del promotor viral LTR, produciéndose alteraciones en el crecimiento y diferenciación celular.
- Translocación o reordenación cromosómica: Es el cambio de localización de una porción cromosómica, con los genes que lleva incorporados a otra ubicación distinta dentro del mismo cromosoma o de otro, que pueden afectar a la expresión o función bioquímica de un protooncogén. Las translocaciones ocurren frecuentemente en los tumores hematológicos como los linfomas y leucemias. Por ejemplo el protooncogén c-myc está situado en el cromosoma 8 y puede trasladarse al cromosoma 14. Esta nueva posición produce una sobreexpresión de la proteína que codifica, dando lugar al linfoma de Burkitt. También la leucemia mieloide crónica se produce por la translocación recíproca entre el cromosoma 9 y 22, produciéndose un oncogén híbrido entre el gen c-abl del cromosoma 9 y la región bcr del cromosoma 22, dando lugar al cromosoma Philadelphia.
- Amplificación: Es el aumento del número de copias del mismo protooncogén del genoma, incluso varias miles de veces. Los cromosomas de los tumores con oncogenes amplificados poseen trastornos estructurales que se visualizan fácilmente en el cariotipo como regiones con bandas anómalas, regiones teñidas homogéneamente (Homogeneously Staining Regions, HSR en inglés) o “diminutos dobles” (double minutes, DM en inglés) que son pequeños fragmentos extracromosómicos de tamaño variable que se replican automáticamente. En varios tumores se ha detectado amplificación oncogénica y el grado de amplificación está muy relacionado en el estadio y pronóstico del tumor. La sobreexpresión por amplificación del oncogén n-myc produce el neuroblastoma, aunque también se encuentra en otros tumores. El aumento del número de copias de un oncogén además de producir un aumento de la proteína que codifica y que actúa como factor de crecimiento, también produce un mayor aumento de receptores al factor de crecimiento. Los protooncogenes amplificados en los tumores humanos pertenecen sobre todo a una de estas tres familias: erb B, ras o myc. Se desconoce todavía si la amplificación protooncogénica es causa o produce malignidad en un tumor, por ejemplo para que adquiera la capacidad de metastatizar o es un efecto de la transformación maligna de un tumor ya que ocurre en tumores grandes, poco diferenciados y que tienen metástasis, cualidades que aumentan la probabilidad de transformación maligna de la amplificación.
- Hipometilación: Se estima que entre un 2 y un 7% de los residuos de citosina en el ADN están metilados. Cuando los grupos metilo (CH3) se localizan en secuencias de ADN promotoras de genes, la iniciación de la transcripción se encuentra mecánicamente interferida, siendo el grado de transcripción inversamente proporcional a la metilación. La disminución de grupos metilo en las bases de citosina de una secuencia promotora de un protooncogén, activa su transcripción y la posible transformación maligna a un oncogén.
Mecanismos cualitativos
- Mutación puntual: La sustitución de una base nitrogenada en el ADN de un gen, puede producir un cambio en el aminoácido identificado por el codón que presenta la mutación, que provoca un cambio estructural en la proteína sintetizada por ese gen, alterándose su función, por lo que la sustitución de una sola base nitrogenada en la cadena de ADN puede transformar un protooncogén en un oncogén. Por ejemplo el oncogén ras modifica un codón de lectura que convierte la glicina en valina. Oncogenes homólogos como el H-RAS, K-RAS Y N-RAS también poseen mutaciones puntuales en otras localizaciones. Los puntos donde se producen dichas mutaciones son críticos para el control del crecimiento celular normal, ya que en el caso del oncogén ras, las mutaciones impiden la conversión de la forma activa a inactiva, con la consiguiente alteración en el control de la proliferación celular.
- Deleción del material genético: La pérdida de material genético de un cromosoma puede activar a un oncogén por medio de tres mecanismos:
- La pérdida puede ser de una secuencia inhibitoria de un protooncogén, que provoca la sobreexpresión del producto del oncogén.
- La pérdida puede provocar que el oncogén quede más cerca de una secuencia promotora, produciendo también una sobreexpresión.
- La pérdida puede ser de un gen supresor tumoral, y suele ser el mecanismo probablemente más importante por el que una pérdida cromosómica puede activar un oncogén.
Proteínas codificadas por los oncogenes
Los protooncogenes codifican las proteínas necesarias que intervienen en el control de la mayor parte de los mecanismos por los que se regula la proliferación celular como:
- Factores de crecimiento.
- Receptores de factores de crecimiento.
- Receptores hormonales.
- Factores de transmisión intracelular o segundos mensajeros como:
- Proteínas con actividad tirosinquinasa.
- Proteínas de unión a guanosina trifosfato.
- Factores de transcripción nuclear.
Tipos de oncogenes
| Nombre | Función del oncogen | Tumor | |||
|---|---|---|---|---|---|
| abl | tirosina kinasa | leucemia mieloide crónica | |||
| erb-B | receptor EGF (tirosina kinasa) | carcinoma espinocelular | |||
| fes | tirosina kinasa | sarcoma | |||
| fms | receptor de M-CSF, tirosina kinasa | sarcoma | |||
| fos, jun | los productos se asocian para formar una prot. reg. de AP-1 | osteosarcoma, sarcoma | |||
| kit | receptor factor Steel, tirosina kinasa | sarcoma | |||
| raf | serina/treonina kinasa, activada por Ras | sarcoma | |||
| L-myc | factor de transcripción | cáncer de pulmón | |||
| N-myc | factor de transcripción | neuroblastoma | |||
| Neu | neuroblastoma, cáncer de mama | ||||
| H-Ras | proteína que une GTP | cáncer de vejiga y cáncer de riñón | |||
| K-Ras | proteína que une GTP | cáncer de páncreas, cáncer de colon | N-Ras | proteína que une GTP | Melanomas, tumores malignos hematológicos |
| rel | proteína reguladora relacionada con NFkappaΒ | reticuloendoteliosis | |||
| ret | cáncer de tiroides | ||||
| sis | cadena B del PDGF | sarcoma | |||
| src | proteína kinasa | sarcoma, cáncer de colon |
Historia de los oncogenes
El inicio del descubrimiento de los oncogenes fue a través de los estudios del patólogo Francis Peyton Rous que trabajaba en el Instituto Rockefeller de Nueva York en 1910. Rous transmitió el sarcoma de pollo, mediante la inyección a docenas de gallinas de un extracto de cultivo celular tumoral, que no contenía células vivas. Con este procedimiento consiguió reproducir el tumor y sospechó que el agente causal debería ser de menor tamaño que las células y que las bacterias, por lo que podría corresponder a un virus, aunque no lo llamó así, sino agente carcinógeno. Más tarde descubrió que era un virus y por sus descubrimientos le concedieron el premio Nobel de Medicina en 1966.
El virus del sarcoma de Rous (src) es el prototipo de los retrovirus, demostrando que la información genética no se transfiere solamente de forma unidireccional de ADN a ARN y de este a proteínas, sino que los retrovirus mediante la enzima transcriptasa inversa, son capaces de sintetizar ADN a partir de ARN. Además de descubrir que el src era carcinogénico, se descubrió que estaba formado por cuatro genes, tres de los cuales imprescindibles para la multiplicación del virus y el cuarto gen, el v-src, que no realiza ninguna función en el virus, pero que en las células produce la transformación maligna cuando se infectan por el virus. En 1975 el trabajo de los Dres Ferrer-Roca O. y Egozcue Cuixart J. sobre la importancia de los virus ubicuos desarrollaba la teoría de la correlación cariotipo tumoral dentro de la teoría somatico-viral del cáncer en la que se estipulaba que la inserción de los virus oncogenos provocaban las anomalías cromosómicas de los tumores y luego sufrían un proceso de selección clonal. En 1989 John Michael Bishop, que también recibió el premio Nobel de Medicina en 1989, descubrió mediante técnicas de ingeniería genética que las secuencias homólogas del v-src también se encuentran en el ADN de las células normales, tanto de aves como de muchos vertebrados e incluso en el ser humano, demostrando así que el oncogén src procede de un gen normal animal, un protooncogén, por lo que la transformación cancerosa se produce en genes normales, los protooncogenes que realizan funciones muy importantes en la proliferación y diferenciación celular. El estudio de la secuencia de nucleótidos del v-src, al poseer intrones y exones, que son secuencias exclusivas de ADN animales y no virales, se dedujo que este gen no pertenece al virus, sino que debió ser arrastrado por el virus después de unirse y desprenderse del ADN de alguna célula animal infectada durante la evolución. El protooncogén c-src, de las células normales codifica una proteína llamada por Bishop pp60c-src que está muy unida a la superficie interior de la membrana celular, capaz de fosforilar las tirosinas. Se sabe que las células cancerosas que contienen el oncogén v-src activo, presentan una proteína pp60c-src anormal, a la que le falta un residuo de tirosina en un lugar concreto de su conformación y que en la versión normal de la proteína está fosforilada para bloquear la fosforilación de otras proteínas en la transmisión de señales. Por eso la pp60c-src mutada está continuamente funcionando, añadiendo grupos fosfato en estas proteínas de transmisión de señales en las células cancerosas.
La demostración final de que el cáncer es una enfermedad genética se remonta a principio de los años ochenta, gracias a Robert A.Weinberg, Geofrey M. Cooper, Michael Wigler y Mariano Barbacid pertenecientes a equipos de investigación diferentes. Estos cuatro científicos aislaron muestras de ADN de diferentes tumores humanos y lo añadieron a cultivos celulares de fibroblastos no tumorales de ratón. Las células fibroblásticas normales dejan de multiplicarse cuando entran en contacto unas con otras, fenómeno denominado inhibición por contacto, mientras que grupos celulares de los cultivos de fibroblastos que contenían ADN tumoral se multiplicaban sin control, perdiendo la inhibición por contacto y además formaban nódulos tumorales al inocularlos en ratones inmunodeprimidos, fenómeno que no ocurría al inyectar fibroblastos normales. Se volvieron a aislar las células fibroblásticas transformadas en tumorales, fragmentando su ADN mediante enzimas de restricción, y los fragmentos obtenidos de ADN se inyectaron de nuevo en fibroblastos normales, volviéndose unos cancerosos y otros no. El proceso de aislar a las células tumorales, fragmentar su ADN e inocularlo a células normales se repitió varias veces, hasta aislar cada vez más fragmentos de ADN humano que provoca cáncer, es decir los oncogenes.
Véase también
Referencias
- Alberts et al (2004). «Biología molecular de la célula». Barcelona: Omega. ISBN 54-282-1351-8.
- Weinberg, R.A. (1996). «How cancer arises». Scientific American (New York: Munn \& Co.) 275 (3): 62-71.