Punto de restricción
El punto de restricción (R) es un punto en G1 del ciclo celular animal en el que la célula se "compromete" con el ciclo celular y después del cual ya no se requieren estimulantes de proliferación extracelular.[1]
| Ciclo celular | |
|---|---|
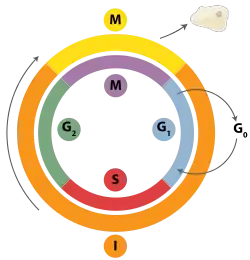 | |
| Interfase | |
| Fase G1 / Fase S / Fase G2 | |
| División celular | |
| Puntos de control | |
| |
| Otras fases celulares | |
| Ciclina | |
| CDK | |
| Inhibidor de CDK | |
| Otras proteínas del ciclo celular | |
Historia
Originalmente, Howard Martin Temin demostró que las células de pollo alcanzan un punto en el que se comprometen a replicar su ADN y no dependen de las señales extracelulares.[2] Cerca de 20 años después, en 1973, Arthur Pardee demostró que existe un único punto de restricción en G1. Anteriormente, G1 se había definido simplemente como el tiempo entre la mitosis y la fase S . No se conocían lugar-marcadores moleculares o morfológicos para la posición de una celda en G1. Pardee utilizó un método de doble bloque en el que cambió las células de un bloque del ciclo celular (como la extracción crítica de aminoácidos o la extracción de suero) a otra y comparó la eficiencia de cada bloque para prevenir la progresión a la fase S. Encontró que ambos bloques en todos los casos examinados fueron igualmente eficientes para bloquear la progresión de la fase S, lo que indica que todos deben actuar en el mismo punto en G1, al que denominó "punto de restricción", o punto - R.[3]
En 1985, Zetterberg y Larsson descubrieron que, en todas las etapas del ciclo celular, la privación de suero produce inhibición de la síntesis de proteínas. Solo en las células postmitóticas (es decir, células en primeros G1) células fuerza de retirada de suero lo hicieron en quiescencia (G0). De hecho, Zetterberg descubrió que prácticamente toda la variabilidad en la duración del ciclo celular se puede tener en cuenta en el tiempo que tarda la célula en moverse desde el punto de restricción hasta la fase S.[4]
Señales extracelulares
Excepto por el desarrollo embrionario temprano, la mayoría de las células en los organismos multicelulares persisten en un estado de reposo conocido como G0, donde no ocurre la proliferación, y las células típicamente se diferencian terminalmente; otras células especializadas continúan dividiéndose en la edad adulta. Para ambos grupos de células, se tomó la decisión de salir del ciclo celular y quedarse inactivo (G0), o volver a ingresar a G1.
La decisión de una célula de ingresar o reingresar al ciclo celular se realiza antes de la fase S en G1 en lo que se conoce como el punto de restricción, y está determinada por la combinación de señales extracelulares promocionales e inhibitorias que se reciben y procesan. Antes del punto R, una célula requiere que estos estimulantes extracelulares comiencen a progresar a través de las tres primeras subfases de G1 (competencia, entrada G1a, progresión G1b). Sin embargo, después de que se haya pasado el punto R en G 1b, ya no se requieren señales extracelulares y la célula se compromete irreversiblemente a prepararse para la duplicación de ADN. La progresión adicional está regulada por mecanismos intracelulares. La eliminación de estimulantes antes de que la célula alcance el punto R puede provocar la reversión de la célula a la inactividad.[1][2] En estas condiciones, las células realmente se vuelven a establecer en el ciclo celular, y requerirán un tiempo adicional (aproximadamente 8 horas más que el tiempo de extracción en el cultivo) después de pasar el punto de restricción para ingresar a la fase S.[2]
Señalización mitógena
Los factores de crecimiento (por ejemplo, PDGF , FGF y EGF) regulan la entrada de células en el ciclo celular y la progresión al punto de restricción. Después de pasar este "punto de no retorno" similar a un interruptor, la finalización del ciclo celular ya no depende de la presencia de mitógenos.[5][3][6] La señalización mitógena sostenida promueve la entrada en el ciclo celular en gran medida a través de la regulación de las ciclinas G1 (ciclina D1-3) y su ensamblaje con Cdk4/6, que puede mediarse en paralelo a través de las vías MAPK y PI3K.
MAPK señalización en cascada
La unión de los factores de crecimiento extracelular a sus tirosina quinasas receptoras (RTK) desencadena un cambio conformacional y promueve la dimerización y la autofosforilación de los residuos de tirosina en la cola citoplasmática de las RTK. Estos residuos tirosina fosforilados facilitan el acoplamiento de proteínas que contienen un dominio SH2 (por ejemplo, Grb2 ), que posteriormente pueden reclutar otras proteínas de señalización a la membrana plasmática y desencadenar cascadas de quinasa de señalización. Grb2 asociado a RTK se une a Sos , que es un factor de intercambio de nucleótidos de guanina que convierte Ras unido a la membrana en su forma activa (Ras-GDP Ras-GTP).[7] Active Ras activa la cascada de la MAP quinasa, uniendo y activando Raf, que fosforila y activa la MEK, que fosforila y activa la ERK (también conocida como MAPK).

La ERK activa luego se traslada al núcleo donde activa múltiples objetivos, como el factor de transcripción respuesta en suero (SRF), lo que da como resultado la expresión de genes tempranos inmediatos, en particular los factores de transcripción Fos y Myc.[7][8] Los dímeros Fos/Jun comprenden el complejo de factor de transcripción AP-1 y activan los genes de respuesta retardada, incluida la ciclina G1 principal, la ciclina D1.[7] Myc también regula la expresión de una amplia variedad de genes pro-proliferativos y pro-crecimiento, incluyendo alguna inducción de ciclina D2 y Cdk4.[4] Además, la actividad de ERK sostenida parece ser importante para la fosforilación y la localización nuclear de CDK2,[7] además de apoyar la progresión a través del punto de restricción.
Señalización de vía PI3K
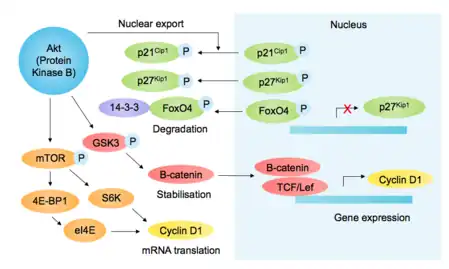
p85, otra proteína que contiene el dominio SH2, se une a las RTK activadas y recluta PI3K (fosfoinositido-3-quinasa), fosforilando el fosfolípido PIP2 a PIP3, lo que lleva al reclutamiento de Akt (a través de su dominio PH). Además de otras funciones pro-crecimiento y pro-supervivencia, Akt inhibe la glucógeno sintasa quinasa-3β (GSK3β), previniendo así la fosforilación mediada por GSK3β y la posterior degradación de la ciclina D1 [9] ( ver figura [10] ). Akt además regula los componentes G1/S mediante la promoción mediada por mTOR de la traducción de ciclina D1,[11] fosforilación de los inhibidores de Cdk p27 kip1 (evitando su importación nuclear) y p21 Cip1 (estabilidad decreciente), e inactivando la fosforilación del factor de transcripción FOXO4 (que regula la expresión de p27).[12] En conjunto, esta estabilización de la ciclina D1 y la desestabilización de los inhibidores de Cdk favorecen la actividad de G1 y G1/S-Cdk.
Señalización anti mitogénica
Los anti-mitógenos como la citoquina TGF-β inhiben la progresión a través del punto de restricción, causando una detención de G1. La señalización de TGF-β activa Smads, que se compleja con E2F4/5 para reprimir la expresión de Myc y también se asocia con Miz1 para activar la expresión del inhibidor de Cdk p15 INK4b para bloquear la formación y actividad del complejo de ciclina D-Cdk.[7][13] Las células detenidas con TGF-β también acumulan p21 y p27.[14]
Mecanismo
Visión general
Como se describió anteriormente, las señales de los factores de crecimiento extracelular se transducen de una manera típica. El factor de crecimiento se une a los receptores en la superficie celular, y una variedad de cascadas de fosforilación dan como resultado la captación de Ca 2+ y la fosforilación de proteínas. Los niveles de fosfoproteínas están compensados por las fosfatasas. En última instancia, se produce la activación transcripcional de ciertos genes diana. La señalización extracelular debe mantenerse, y la célula también debe tener acceso a suficientes suministros de nutrientes para apoyar la síntesis rápida de proteínas. La acumulación de ciclina D es esencial.[15]
Los cdks 4 y 6 unidos a la ciclina D se activan mediante la quinasa activadora de cdk y conducen la célula hacia el punto de restricción. La ciclina D, sin embargo, tiene una alta tasa de rotación (t1/2<25 min). Debido a esta rápida tasa de rotación, la célula es extremadamente sensible a los niveles de señalización mitogénica, que no solo estimulan la producción de ciclina D, sino que también ayudan a estabilizar la ciclina D dentro de la célula.[15][16] De esta manera, la ciclina D actúa como un sensor de señal mitogénica.[16] Los inhibidores de Cdk (CKI), como las proteínas Ink4 y p21 , ayudan a prevenir la actividad inadecuada de ciclina-cdk.
Los complejos activos de ciclina D-cdk fosforilan la proteína del retinoblastoma (pRb) en el núcleo. La Rb no fosforilada actúa como un inhibidor de G1 al prevenir la transcripción mediada por E2F. Una vez fosforilada, E2F activa la transcripción de las ciclinas E y A.[15][16][17] La ciclina E-cdk activa comienza a acumularse y completa la fosforilación de pRb, como se muestra en la figura.[18]
Inhibidores de Cdk y regulación de la actividad del complejo Ciclina D / Cdk
p27 y p21 son inhibidores estequiométricos de los complejos G1/S y S-ciclina-Cdk. Mientras que los niveles de p21 aumentan durante la entrada en el ciclo celular, la p27 generalmente se desactiva a medida que las células avanzan hacia el final de G1.[7] La alta densidad celular, la inanición de mitógenos y el TGF-β dan como resultado la acumulación de p27 y la detención del ciclo celular.[14] De manera similar, el daño en el ADN y otros factores estresantes aumentan los niveles de p21, mientras que la actividad de ERK2 y Akt estimulada por mitógenos conduce a la inactivación de la fosforilación de p21.[19]
El trabajo inicial sobre la sobreexpresión de p27 sugirió que puede asociarse con e inhibir los complejos de ciclina D-Cdk4 / 6 y los complejos de ciclina E / A-Cdk2 in vitro y en tipos de células seleccionadas.[14] Sin embargo, los estudios cinéticos de LaBaer et al. (1997) encontraron que la titulación en p21 y p27 promueve el ensamblaje del complejo ciclina d-Cdk, aumentando la actividad general y la localización nuclear del complejo.[20] Estudios posteriores demostraron que p27 puede ser necesario para la formación del complejo ciclina D-Cdk, ya que p27 - / - , p21 - / - MEF mostraron una disminución en la complejación de ciclina D-Cdk4 que podría ser rescatada con la expresión de p27.[21]
El trabajo de James et al. (2008) sugiere además que la fosforilación de los residuos de tirosina en p27 puede cambiar p27 entre un estado inhibitorio y no inhibitorio mientras se une a la ciclina D-Cdk4 / 6, ofreciendo un modelo de cómo p27 es capaz de regular el ensamblaje del complejo ciclina-Cdk y actividad.[22] La asociación de p27 con ciclina D-Cdk4 / 6 puede promover aún más la progresión del ciclo celular al limitar el conjunto de p27 disponible para inactivar los complejos de ciclina E-Cdk2.[7][23] El aumento de la actividad de ciclina E-Cdk2 en la G1 tardía (y la ciclina A-Cdk2 en la S temprana) conduce a la fosforilación de p21 / p27 que promueve su exportación nuclear, ubiquitinación y degradación.
Dinámica
Un artículo publicado por los grupos Lingchong You y Joe Nevins en la Universidad de Duke en 2008 demostró que el interruptor histérico biestable E2F subyace en el punto de restricción. E2F promueve su propia activación, y también promueve la inhibición de su propio inhibidor ( pRb ), formando dos circuitos de retroalimentación (entre otros) que son importantes para establecer sistemas biestables. Los autores de este estudio utilizaron un sistema GFP desestabilizado bajo el control del promotor E2F como lectura de la actividad E2F. Las células privadas de suero se estimularon con concentraciones séricas variables, y la lectura de GFP se registró a nivel de una sola célula. Encontraron que el reportero de GFP estaba encendido o apagado, lo que indicaba que E2F estaba completamente activado o desactivado en todos los diferentes niveles de suero analizados. Otros experimentos, en los que analizaron la dependencia histórica del sistema E2F, confirmaron que funciona como un interruptor biestable histerético.[24]
En el cáncer
El cáncer se puede ver como una interrupción de la función del punto de restricción normal, ya que las células vuelven a ingresar de manera continua e inapropiada en el ciclo celular y no ingresan a G0.[1] Las mutaciones en muchos pasos en el camino hacia el punto de restricción pueden resultar en un crecimiento canceroso de las células. Algunos de los genes más comúnmente mutados en el cáncer incluyen Cdks y CKI; los Cdks hiperactivos o los CKI inactivos disminuyen la rigurosidad del punto de restricción, lo que permite que más células eviten la senescencia.[17]
El punto de restricción es una consideración importante en el desarrollo de nuevas terapias con medicamentos. En condiciones fisiológicas normales, toda la proliferación celular está regulada por el punto de restricción. Esto puede ser explotado y utilizado como una forma de proteger las células no cancerosas de los tratamientos de quimioterapia. Los medicamentos de quimioterapia típicamente atacan las células que están proliferando rápidamente. Al usar medicamentos que inhiben la terminación del punto de restricción, como los inhibidores del receptor del factor de crecimiento , se evita que las células normales proliferen y, por lo tanto, se protegen de los tratamientos de quimioterapia.[16]
Referencias
- Pardee, A. (1989). «G1 events and regulation of cell proliferation». Science 246 (4930): 603-8. Bibcode:1989Sci...246..603P. PMID 2683075. doi:10.1126/science.2683075.
- Zetterberg, Anders; Larsson, Olle; Wiman, Klas G (1995). «What is the restriction point?». Current Opinion in Cell Biology 7 (6): 835-42. PMID 8608014. doi:10.1016/0955-0674(95)80067-0.
- Pardee, Arthur B. (1974). «A Restriction Point for Control of Normal Animal Cell Proliferation». Proceedings of the National Academy of Sciences 71 (4): 1286-90. Bibcode:1974PNAS...71.1286P. PMC 388211. PMID 4524638. doi:10.1073/pnas.71.4.1286.
- Zetterberg, A.; Larsson, Olle (1985). «Kinetic Analysis of Regulatory Events in G1 Leading to Proliferation or Quiescence of Swiss 3T3 Cells». Proceedings of the National Academy of Sciences 82 (16): 5365-9. Bibcode:1985PNAS...82.5365Z. PMC 390569. PMID 3860868. doi:10.1073/pnas.82.16.5365.
- Blagosklonny, M.V. and Pardee, A.B. (2002). «The restriction point of the cell cycle». Cell Cycle 1: 102-109.
- Pardee, Arthur B. (1 de abril de 1974). «A Restriction Point for Control of Normal Animal Cell Proliferation». Proceedings of the National Academy of Sciences (en inglés) 71 (4): 1286-1290. ISSN 1091-6490. PMID 4524638. doi:10.1073/pnas.71.4.1286.
- Morgan, D.O. (2007). The Cell Cycle: Principles of Control. New Science Press. pp. 208–213.
- Adhikary, S. and Eilers, M. (2005). «Transcriptional regulation and transformation by Myc proteins». Nat. Rev. Mol. Cell 1: 102-109.
- Diehl, J. (1998). «Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization». Genes Dev. 12: 3499-3511.
- VanArsdale, T. (2015). «Targeting the cyclin D-Cdk4/6 axis for cancer treatment». Clin. Cancer Res. 21: 2905-2910.
- Hay, N. and Sonenberg, N. (2004). «Upstream and downstream of mTOR». Genes Dev. 18: 1926-1945.
- Lu, Z., and Hunter, T. (2010). «Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors». Cell Cycle 9: 2342-2352.
- Shi, Y., and Massague, J. (2003). «Mechanisms of TGF-β signaling from cell membrane to the nucleus». Cell 113: 685-855.
- Denicourt, C. and Dowdy, S.F. (2004). «Mechanisms of TGF-β signaling from cell membrane to the nucleus». Genes Dev. 8: 851-855.
- Sherr, Charles J.; Roberts, James M. (1995). «Inhibitors of mammalian G1 cyclin-dependent kinases». Genes & Development 9 (10): 1149-63. PMID 7758941. doi:10.1101/gad.9.10.1149.
- Blagosklonny, Mikhail V.; Pardee, Arthur B. (2001). «The Restriction Point of the Cell Cycle». En Blagosklonny, Mikhail V., ed. Cell Cycle Checkpoints and Cancer. Austin: Landes Bioscience. pp. 52–?. ISBN 978-1-58706-067-0.
- Malumbres, Marcos; Barbacid, Mariano (2001). «Milestones in Cell Division to Cycle or Not to Cycle: A Critical Decision in Cancer». Nature Reviews Cancer 1 (3): 222-31. PMID 11902577. doi:10.1038/35106065.
- Holsberger, Denise R.; Cooke, Paul S. (2005). «Understanding the role of thyroid hormone in Sertoli cell development: A mechanistic hypothesis». Cell and Tissue Research 322 (1): 133-40. PMID 15856309. doi:10.1007/s00441-005-1082-z.
- Sherr, C.J. and Roberts, J.M. (1999). «CDK inhibitors: positive and negative regulators of G1-phase progression». Genes Dev. 13: 1501-1512.
- LaBaer, J. (1997). «New functional activities for the p21 family of CDK inhibitors». Genes Dev. 11: 847-862.
- Cheng, M. (1999). «The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts». EMBO J. 18: 1571-1583.
- James, M.K. (2008). «Differential modification of p27Kip1 controls its cyclin D-Cdk4 inhibitory activity». Mol. Cell. Biol. 28: 498-510.
- Goel, S. (2018). «CDK4/6 inhibition in cancer: beyond cell cycle arrest». Trends Cell Biol. 28: 911-925.
- Yao, Guang; Lee, Tae Jun; Mori, Seiichi; Nevins, Joseph R.; You, Lingchong (2008). «A bistable Rb–E2F switch underlies the restriction point». Nature Cell Biology 10 (4): 476-82. PMID 18364697. doi:10.1038/ncb1711.
| Control de autoridades |
|
|---|
 Datos: Q7316321
Datos: Q7316321