Punto de control de la mitosis
El punto de control de la mitosis o punto de control del ensamblaje del huso (SAC), asegura que la segregación cromosómica tenga lugar de forma correcta. La comprensión de cómo las células mantienen constante el número apropiado de cromosomas después de cada división celular es una cuestión central en un sinnúmero de procesos biológicos. Un único error que dé lugar a células con más o menos cromosomas de los que deber tener (una situación denominada aneuploidía), puede conducir en el mejor de los casos a la muerte celular, o bien producir resultados fenotípicos problemáticos:
- En humanos, el síndrome de Down aparece en niños que portan en sus células un cromosoma 21 en exceso, como resultado de un fallo en la segregación cromosómica durante la meiosis en uno de sus progenitores. Ese fallo dará lugar a un gameto (espermatozoide u óvulo) con un cromosoma 21 extra, que tras la fecundación generará un embrión que consecuentemente recibirá el cromosoma excedente.
- En las células cancerosas, la aneuploidía es relativamente frecuente, lo que indica que estas células presentan algún defecto en la maquinaria implicada en la segregación cromosómica y en el mecanismo que asegura que dicha segregación ocurre de forma correcta.
| Ciclo celular | |
|---|---|
 | |
| Interfase | |
| Fase G1 / Fase S / Fase G2 | |
| División celular | |
| Puntos de control | |
| |
| Otras fases celulares | |
| |
| Ciclina | |
| CDK | |
| Inhibidor de CDK | |
| Otras proteínas del ciclo celular | |
Los mecanismos que verifican que se cumplen los requisitos necesarios para pasar a la fase siguiente del ciclo celular se denominan puntos de control (checkpoints). A través del ciclo celular, existen diferentes puntos de control. El punto de control que asegura que la segregación cromosómica tiene lugar de forma correcta se denomina punto de control de la mitosis, de anafase o también punto de control del ensamblaje del huso, abreviado SAC por sus siglas en inglés (Spindle Assembly Checkpoint).
Segregación cromosómica
División celular: duplicación de material y distribución a las células hijas
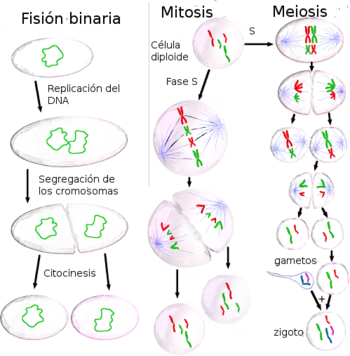
Cuando las células están preparadas para dividirse, bien porque el tamaño celular es lo bastante grande o porque reciben el estímulo apropiado,[1] activan los mecanismos de entrada en el ciclo celular y duplican la mayoría de sus orgánulos durante la fase S (de síntesis), incluido su centrosoma, de manera que cuando finalice el proceso de división cada célula hija recibirá un juego completo de orgánulos. Asimismo, durante la fase S todos los organismos deben duplicar su ADN con precisión, un proceso conocido como replicación de ADN. Una vez terminada la replicación, en los organismos eucariotas el ADN se compacta y se condensa, formando los cromosomas mitóticos, que constan cada uno de dos cromátidas hermanas; cada cromátida es una molécula de ADN completa, unida mediante microtúbulos a uno de los dos centrosomas de la célula en división, localizados en los polos opuestos de la célula. La estructura que forman los centrosomas y los microtúbulos se conoce como huso mitótico, debido a su forma característica, con los cromosomas unidos entre los dos centrosomas. Ambas cromátidas hermanas permanecen unidas hasta anafase, momento en el que se separan y se agrupan alrededor de su centrosoma, de forma que al final de la división celular, cada célula hija recibirá un juego completo de cromátidas. El mecanismo por el cual se produce la correcta distribución de cromátidas durante la división celular se denomina segregación cromosómica.
Para asegurar que la segregación cromosómica tiene lugar correctamente, las células han desarrollado un mecanismo preciso y complejo. En primer lugar, las células deben coordinar la duplicación del centrosoma con la replicación del ADN, y un fallo en esta coordinación producirá inevitablemente la formación de husos monopolares o multipolares, que generalmente provocarán una segregación cromosómica anormal,[2] dado que en este caso, los cromosomas no se distribuirán de forma equilibrada entre las células hijas.
Mitosis: anclaje de los cromosomas al huso y segregación
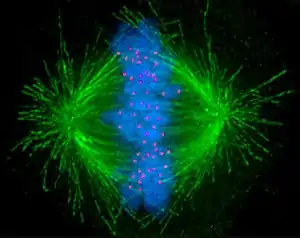
Durante la fase de síntesis (fase S) del ciclo celular, el centrosoma comienza a duplicarse. Justo al inicio de mitosis, ambos centriolos de cada centrosoma alcanzan su longitud máxima, los centrosomas reclutan material adicional y su capacidad de nucleación de microtúbulos aumenta. A medida que progresa la mitosis, ambos centrosomas se separan para establecer el huso mitótico.[3] De esta forma, el huso de una célula mitótica tiene dos polos que emanan microtúbulos. Los microtúbulos son largos filamentos proteicos con dos extremos asimétricos, un extremo "menos" (-) relativamente estable cercano al centrosoma, y un extremo "más" (+) que sufre fases alternadas de crecimiento-retroceso y que explora el centro celular. Cada cromátida presenta una región especial, el centrómero, sobre la que se ensambla una estructura proteica denominada cinetocoro, capaz de estabilizar microtúbulos. Por tanto, si por azar un microtúbulo encuentra un cinetocoro cuando explora el centro celular, puede ocurrir que el cinetocoro lo capture, de forma que el cromosoma queda enganchado al huso mitótico a través del cinetocoro de una de sus cromátidas hermanas. Como los cromosomas presentan dos cinetocoros asociados espalda-con-espalda (uno en cada cromátida hermana), cuando uno de ellos se engancha a los microtúbulos generado por uno de los polos celulares, el cinetocoro de la cromátida hermana queda expuesto hacia el otro polo celular, por lo que en la mayor parte de los casos el segundo cinetocoro se asocia a los microtúbulos del polo opuesto,[4] de manera que los cromosomas quedan « bi-orientados », una configuración fundamental (también denominada anfitélica) para asegurar que la segregación tendrá lugar de manera correcta cuando la célula se divida.[5][6] En ocasiones, uno de los dos cinetocoros hermanos puede anclarse simultáneamente a MTs generados por ambos polos, una configuración denominada merotélica, que no se detecta por el punto de control de la mitosis, pero puede generar cromosomas retrasados en anafase y consecuentemente aneuploidía. La orientación merotélica (que se caracteriza por no presentar tensión entre cinetocoros hermanos) es frecuente al inicio de mitosis, pero la proteína Aurora B (una kinasa conservada desde levaduras hasta vertebrados) detecta y elimina este tipo de anclaje.[7] (Nota: Aurora B se encuentra sobreexpresada frecuentemente en varios tipos de tumores y es en la actualidad una diana para el desarrollo de drogas anticancerosas[8]).
Los cromosomas enganchados a los microtúbulos a través de los cinetocoros son bastante móviles durante la primera parte de la metafase (la ‘prometafase’), oscilando de forma alternada entre ambos polos. El movimiento está estrechamente acoplado con las reacciones de ensamblaje y disociación de los extremos “más” de los microtúbulos, enganchados a los cinetocoros. En un momento dado, todos los cromosomas se alinean en el medio de la zona central del huso (un proceso denominado congregación cromosómica), formando la denominada placa metafásica.[9][10] La maquinaria celular que provoca la degradación de las proteínas que mantienen unidas las cromátidas hermanas (las cohesinas; ver la sección Cohesión de cromátidas hermanas durante mitosis) se activa sólo cuando todos los cromosomas se encuentran alineados en la placa metafásica, disparando la entrada en anafase. De esta manera se asegura que ningún cromosoma se queda completo en una de las células hijas, lo que daría lugar a una célula aneuploide. Dado que cada cromátida hermana está enganchada a uno de los polos, cuando las dos células hijas se reorganicen alrededor de su centrosoma, cada una de ellas recibirá un juego completo de información genética.
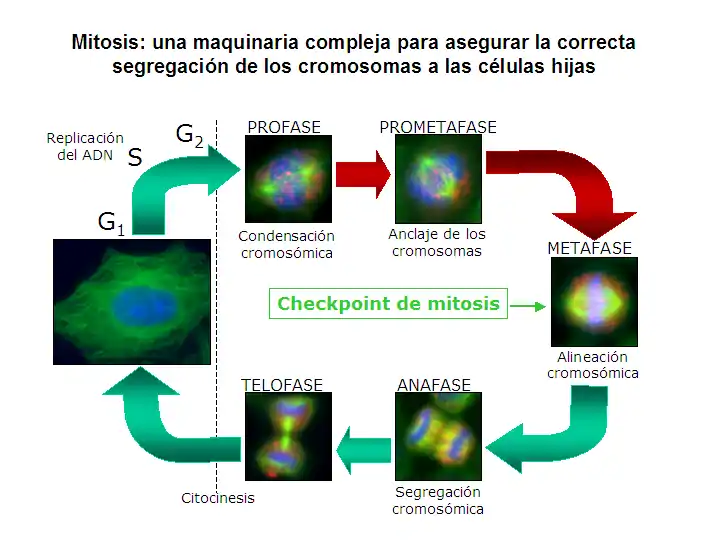
El mecanismo que detecta que se ha formado correctamente un huso mitótico, que todos los cromosomas están asociados a dicho huso de manera bipolar, y que todos ellos se encuentran alineados en la placa metafásica es el denominado punto de control de la mitosis o también punto de control del ensamblaje del huso, abreviado SAC por sus siglas en inglés (Spindle Assembly Checkpoint). Cuando alguno de los cromosomas, por alguna razón, se retrasa durante el proceso de alineamiento, esta maquinaria produce una parada temporal de la progresión en el ciclo celular: la célula se detiene, lo que da tiempo a los mecanismos de reparación para resolver el problema detectado. Si pasado cierto tiempo el problema no se ha corregido, la célula será abocada a un proceso de muerte celular, un mecanismo de seguridad para evitar que se produzca una situación de aneuploidía, generalmente con consecuencias graves para el organismo.
Descubrimiento del punto de control de la mitosis

Uno de los primeros en observar que cuando un cromosoma se retrasa en alcanzar la placa metafásica, el inicio de la anafase se postpone hasta algunos minutos después de su llegada a la placa, fue Zirkle en 1970.[11] Esta observación y otras similares sugerían que existe un mecanismo de control en la transición metafase-anafase. Basándose en el hecho de que utilizando drogas como nocodazol y colchicina se produce un desensamblaje del huso que ocasiona un bloqueo del ciclo celular en metafase (véase la revisión de Rieder y Palazzo en 1992[12]), el mecanismo de control se denominó punto de control del ensamblaje del huso (Spindle Assembly Checkpoint o SAC). Este mecanismo de regulación ha sido el objeto de un intenso estudio desde entonces (véase la revisión de Burke y Stukenberg en 2008[13]).
Diversos estudios genéticos han permitido determinar qué tipo de defectos activan el punto de control de la mitosis: la despolimerización del huso,[14][15] la presencia de cromosomas dicéntricos (con dos centrómeros),[16] centrómeros que segregan de forma aberrante,[17] defectos en los cuerpos de los polos del huso en S. cerevisiae,[18] defectos en las proteínas del cinetocoro,[19] mutaciones en el ADN centromérico[20] o defectos en los motores moleculares activos en mitosis.[14] Un resumen de estas observaciones puede encontrarse en el artículo de Hardwick y colaboradores en 1999.[21]
Basándose en sus propias observaciones, Zirkle[11] fue el primero en proponer que “alguna (…) sustancia, necesaria para que la célula prosiga hacia anafase, aparece algunos minutos después de C (momento de la llegada del último cromosoma a la placa metafásica), o de que ocurra un cambio drástico en las condiciones en el citoplasma justo en el momento C o poco después de C”, sugiriendo que tal función reside en cinetocoros no enganchados al huso. McIntosh extendió esta propuesta, sugiriendo que una enzima sensible a la tensión localizada en los centrómeros produce un inhibidor de la entrada en anafase cuando los dos cinetocoros hermanos no están bajo tensión bipolar.[22] Ciertamente, los datos indicaban que la señal de “espera para entrar en anafase” se producía fundamentalmente en o cerca de los cinetocoros no enganchados al huso.[23] Sin embargo, el suceso primario asociado con el anclaje del cinetocoro al huso, que desactiva la señal inhibidora y libera la parada en metafase, podría ser o bien la adquisición de microtúbulos por el cinetocoro (como proponían Rieder y colaboradores en 1995[23]), o bien la tensión que estabiliza el anclaje de los microtúbulos a los cinetocoros (como sugerían los experimentos realizados en el laboratorio de Nicklas[24]). Estudios posteriores en células que contenían dos husos mitóticos independientes en un único citoplasma mostraron que el inhibidor de la transición metafase-anafase se produce por los cinetocoros no anclados al huso y no se difunde libremente en el citoplasma.[25] Sin embargo, en el mismo estudio se mostraba que una vez que la transición de metafase a anafase se ha iniciado en una parte de la célula, esta información se extiende a través de todo el citoplasma, y puede superar la señal de “espera para entrar en anafase” asociada a un segundo huso que contenga cinetocoros sin anclar.
Cohesión de cromátidas hermanas durante mitosis
Cohesina: las proteínas SMC
Como se ha indicado anteriormente, las cromátidas hermanas de cada cromosoma permanecen asociadas desde la fase S (cuando el ADN se replica para generar dos copias idénticas, las dos cromátidas) hasta anafase, momento en el cual las cromátidas se separan y se dirigen a los polos opuestos de la célula en división. Estudios genéticos y bioquímicos en levaduras y en extractos de huevos de Xenopus laevis identificaron un complejo poliproteínico que juega un papel crítico en la cohesión de las cromátidas hermanas (véase la revisión publicada por Hirano en 2000[26]). Este complejo se conoce como el complejo cohesina y en Saccharomyces cerevisiae está formado por al menos cuatro subunidades: Smc1p, Smc3p, Scc1p (o Mcd1p) y Scc3p. Tanto Smc1p como Smc3p pertenecen a la familia de las proteínas para el mantenimiento estructural de cromosomas (Structural Maintenance of Chromosomes, abreviado SMC), un grupo de ATPasas cromosómicas altamente conservadas, y forman un heterodímero (Smc1p/Smc3p). Scc1p es el homólogo en S.cerevisiae de Rad21, identificada primero como una proteína de reparación del ADN en S. pombe. Estas cuatro proteínas son esenciales en levaduras, y una mutación en cualquiera de ellas produce la separación prematura de las cromátidas hermanas. En levaduras, la cohesina se une a sitios preferenciales a lo largo de los brazos cromosómicos y es muy abundante alrededor de los centrómeros, como se demostró en un ensayo de inmunoprecipitación de cromatina.[27]
El papel de la heterocromatina
Observaciones citológicas clásicas sugerían que las cromátidas hermanas están más fuertemente unidas en las regiones heterocromáticas,[28] lo que condujo a suponer que la estructura o la composición particular de la heterocromatina podría favorecer el reclutamiento de cohesina.[29] De hecho, se ha demostrado que Swi6 (la proteína homóloga de HP-1 en S. pombe) se une a la Lys 9 metilada de la histona H3 y promueve la unión de la cohesina a las repeticiones centroméricas en S. pombe.[30][31] Estudios más recientes revelan que la maquinaria de interferencia de ARN regula el establecimiento de la heterocromatina, que a su vez recluta cohesina a esta región, tanto en S. pombe[32] como en células de vertebrados.[33] Sin embargo, además de la presencia de heterocromatina, deben existir otros mecanismos que aseguren un incremento de cohesión en los centrómeros, dado que, aunque S. cerevisiae carece de heterocromatina próxima al centrómero, la presencia de un centrómero funcional induce un incremento en la asociación de cohesina en una región adyacente que abarca 20-50kb.[34]
En este sentido, Orc2 (una proteína del complejo de reconocimiento de origen —ORC por sus siglas en inglés— implicado en la iniciación de la replicación del ADN durante la fase S) también se localiza en el cinetocoro durante mitosis en células humanas;[35] en concordancia con esta localización, algunos estudios indican que Orc2 en levaduras está implicada en la cohesión de cromátidas hermanas, y su eliminación celular provoca la activación del punto de control de la mitosis.[36] También se ha detectado que otros componentes del complejo ORC (como orc5 en S. pombe) intervienen en cohesión.[37] Sin embargo, la ruta molecular en la que intervienen las proteínas ORC parece ser aditiva a la ruta de las cohesinas y se desconoce en su mayor parte.
Función de la cohesión y su disolución
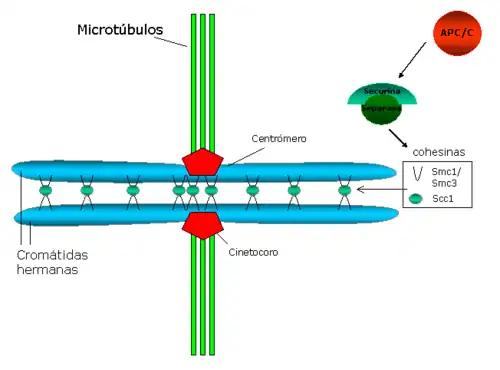
La cohesión centromérica se opone a las fuerzas que ejercen los microtúbulos del huso hacia los polos y que generan tensión entre los cinetocoros hermanos. Esta tensión a su vez estabiliza el anclaje microtúbulo-cinetocoro, a través de un mecanismo que implica la proteína Aurora B (una revisión sobre el tema: Hauf y Watanabe 2004[38]).
Por otro lado, una disminución de los niveles celulares de cohesina causa tanto la separación prematura de cromátidas hermanas como defectos en la alineación de los cromosomas en la placa metafásica, además de la deslocalización del complejo de proteínas pasajeras de los cromosomas (chromosomal passenger complex) que contiene la proteína Aurora B.[39][40] La estructura propuesta del complejo de cohesina sugiere que este complejo conecta directamente ambas cromátidas hermanas.[41] Se ha propuesto que los componentes SMC de la cohesina juegan un papel estructural en la cohesión, de manera que el heterodímero SMC puede funcionar como una molécula que se une al ADN y cuya conformación está regulada por ATP,[42] mientras que Scc1p y Scc3p jugarían un papel regulador.[43]
En S. cerevisiae, la proteína Pds1p (o securina) regula la cohesión entre cromátidas hermanas, al unirse e inhibir la enzima Esp1p (separina o separasa). Cuando se dispara la entrada en anafase, el complejo que promueve anafase (APC/C, las siglas en inglés de Anaphase Promoting Complex o Cyclosome) degrada Pds1p. La degradación de Pds1p/Securina libera la enzima Esp1p/Separasa, que corta Scc1p, lo cual provoca la separación de las cromátidas hermanas.[44] Por otro lado, parece que la kinasa Polo/Cdc5 fosforila residuos de serina adyacentes al sitio de corte de Scc1, lo que facilitaría el corte de esta molécula.[45]
Aunque esta maquinaria está conservada a través de la evolución,[46][47] en vertebrados la mayor parte de las cohesinas se libera en profase, independientemente de la presencia de APC/C, en un proceso dependiente de la kinasa similar a Polo (Plk1) y de Aurora B.[48] Sin embargo, por un lado se ha mostrado que una pequeña cantidad de Scc1 permanece asociada con los centrómeros en células humanas hasta metafase, y una cantidad similar se corta en anafase, cuando desaparece de los centrómeros[49] y por otro algunos experimentos indican que la cohesión entre los brazos cromosómicos se pierde gradualmente después de que los centrómeros hermanos se han separado y las cromátidas hermanas se mueven hacia los polos opuestos de la célula.[50][51]
Ciertas observaciones sugieren que una fracción de las cohesinas de los brazos cromosómicos y las cohesinas centroméricas están protegidas por la proteína Sgo1, que evita que las cohesinas sean desensambladas durante profase.[52][53] Como potencial protector de la cohesión centromérica, la función de Sgo1 tiene que inactivarse al inicio de la anafase, al igual que Pds1p. De hecho, como ocurre con Pds1p, Sgo1 es también un sustrato del APC/C en vertebrados.[54]
Regulación de la progresión del ciclo celular: fosforilación y degradación

La progresión a través del ciclo celular requiere la activación sucesiva de diferentes kinasas dependientes de ciclinas (cyclin-dependent kinases o CDKs), que se regulan mediante la asociación transitoria con subunidades reguladoras (las ciclinas), la unión de polipéptidos inhibidores y fosforilaciones reversibles. Se ha mostrado que muchas proteínas kinasas diferentes juegan un papel importante en la regulación de las diferentes transiciones en el ciclo celular.[55] La iniciación de mitosis está regulada por una cascada de fosforilaciones coordinada espacial y temporalmente que activa el factor de iniciación de mitosis (M-phase/maturation Promoting Factor o MPF) (véase la revisión de Ohi y Gould, 1999[56]). En S. cerevisiae, MPF se compone de la kinasa dependiente de ciclina Cdc28 (Cdc2 en S. pombe o Cdk1 en eucariotas superiores) y como subunidad reguladora una ciclina tipo-B. Como se demostró inicialmente en Schizosaccharomyces pombe, la activación de MPF se centra en el estado de fosforilación del residuo Tyr 15 de Cdc2:[57] MPF se mantiene inactivo en interfase mediante la fosforilación de la Tyr 15 y, en el momento de la entrada en mitosis, MPF se activa mediante la defosforilación de este residuo. La entrada en mitosis se dispara por activación simultánea de la fosfatasa de la Tyr 15 (Cdc25) e inactivación de la kinasa de la Tyr 15, Wee1. La activación de Cdc2 depende en parte de un lazo de retroalimentación positiva, ya que MPF es capaz de fosforilar y por tanto activar y reprimir la actividad de Cdc25 y Wee1, respectivamente. Una vez activado, MPF conduce a las células desde G2 hasta mitosis, mediante la fosforilación de numerosos sustratos, lo que produce la ruptura de la envoltura nuclear, la condensación cromosómica, la separación de los centrosomas, el ensamblaje del huso, la fragmentación del aparato de Golgi y la activación de la maquinaria de segregación cromosómica. La inactivación de MPF (acoplada a la degradación de la ciclina B) facilita el desensamblaje del huso, la decondensación cromosómica, la inactivación de la maquinaria de segregación cromosómica, la citocinesis y la reorganización de la envoltura nuclear, de manera que se generan dos células hijas diferenciadas alrededor del material genético segregado.[55] En levaduras, el inicio de la anafase (definida por la separación de cromátidas hermanas) y la salida de mitosis (indicada por la inactivación de MPF) son dos sucesos clave que requieren la degradación de dos tipos de proteínas:
- la securina (Pds1p, con homólogos en algunos organismos: Cut2 en S. pombe y PTTG en vertebrados).[58]
- las ciclinas mitóticas.[59]
De hecho, células de levaduras que expresan una forma no-degradable de Pds1p se detienen en mitosis con niveles altos de la actividad MPF, lo que indica que la proteólisis de Pds1p es necesaria tanto para la separación de cromátidas hermanas como para la salida de mitosis.[60] Sin embargo, células con deleciones que eliminan la caja de destrucción de la ciclina permanecen en mitosis, aunque las cromátidas hermanas pueden separarse,[61] lo cual indica que la proteólisis de las ciclinas normalmente sigue a la degradación de Pds1p/Securina.
Regulación del APC/C durante la mitosis
Como ya se ha indicado, tanto Pds1p/Securina como las ciclinas son marcadas por el APC/C (Anaphase Promoting Complex o Cyclosome) para su degradación. El APC/C forma parte de un sistema de conjugación por ubiquitina.[62] En general, los sistemas de conjugación de ubiquitina comprenden una enzima activadora (E1) que transfiere la ubiquitina a enzimas conjugadoras de ubiquitina (E2), que en presencia de una ligasa de ubiquitina (E3), unen covalentemente la ubiquitina a la proteína diana, que de esta forma está marcada para su destrucción por el proteasoma. El APC/C es una ligasa E3 específica de mitosis, compuesta de 8-12 subunidades, la mayor parte de ellas conservadas.[63]
Sin embargo, a pesar de esta complejidad, el APC/C necesita una subunidad activadora extra para completar la enzima y promover la ubiquitinación de las proteínas diana. En mitosis existen dos subunidades activadoras:
- Cdc20 en S. cerevisiae (Fizzy en Drosophila y Xenopus laevis, p55Cdc/hCdc20 en humanos, Slp1 en S.pombe)
- Hct1/Cdh1 en S. cerevisiae (Fizzy-relacionada, hCdh1 o Ste9/Srw1 en los organismos correspondientes).
En S. cerevisiae y en humanos, se ha mostrado que tanto Cdc20 como Cdh1 se unen al APC/C, pero a diferencia con las otras subunidades, se unen de forma regulada durante el ciclo celular, no de forma constitutiva.[64] Mutaciones en estas proteínas eliminan la proteólisis dependiente de APC/C.[59][58] La presencia de dos subunidades activadoras del APC/C es muy importante para la regulación del final de la mitosis. En primer lugar, existen evidencias de que Cdc20 y Cdh1 confieren diferente especificidad de substratos sobre el APC/C, y segundo, Cdc20 y Cdh1 están regulados por mecanismos diferentes.
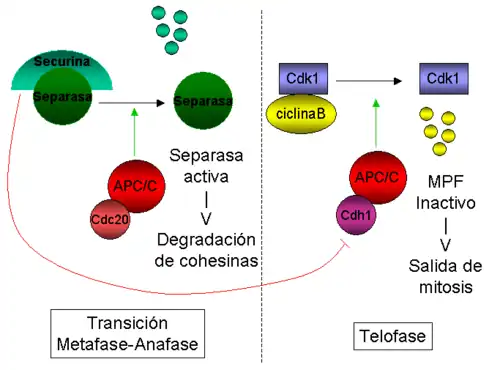
En S. cerevisiae, se ha mostrado que Cdc20 es esencial para la degradación de Pds1/Securina durante la transición de metafase a anafase, mientras que Cdh1 es necesario para la degradación de Clb2 (ciclina B1) más tarde en telofase.[63][59][58] Sin embargo, la diferencia más llamativa entre Cdc20 y Cdh1 no es su especifidad de sustrato, sino su modo de regulación: mientras que la actividad de las CDKs mitóticas parece estimular el APC/C unido a Cdc20 (APC/CCdc20),[65] esta misma actividad inhibe APC/CCdh1.[66]
Por otro lado, en S. cerevisiae el orden correcto de los sucesos mitóticos tardíos se establece en parte mediante mecanismos que aseguran que Cdh1 se activa después de Cdc20. Este orden de sucesos parece basarse en la capacidad de Cdc20 de promover la destrucción de proteínas que inhiben la activación de Cdh1. De hecho, se ha propuesto que la propia Pds1p/Securina es un inhibidor de la destrucción de ciclinas: la sobreexpresión de una forma no-degradable de Pds1p/Securina bloquea no sólo la separación de cromátidas hermanas, sino que además previene la destrucción de las ciclinas y la citocinesis. De acuerdo con estos resultados, hay estudios que muestran que Pds1p/Securina tiene un dobel papel en mitosis:[67] además de secuestrar Esp1p/Separasa, Pdsp1/Securina funciona como un inhibidor de la destrucción de las ciclinas mitóticas, al inhibir la activación de APC/CCdh1. De esta forma, se genera un sistema de seguridad que verifica que las células no saldrán de mitosis hasta que Pds1p/Securina sea destruida y se haya iniciado la anafase.
Sin embargo, además de los cambios secuenciales en la subunidad activadora y en la especificidad de sustrato del APC/C, existen niveles de control suplementarios para asegurar que el orden de los sucesos durante el final de la mitosis tiene lugar de forma correcta:
- La activación del APC/C durante mitosis precisa la fosforilación de Cdc20, Cdh1 y algunas de las subunidades del APC/C.[68]
- La localización subcelular puede proporcionar otro nivel de regulación en la identificación de los sustratos diana del APC/C:
- Las subunidades de APC/C se concentran en los cinetocoros, los polos del huso mitótico y a lo largo de los microtúbulos del mismo.[69]
- Cdc20 (p55CDC ) también se localiza en los cinetocoros a través de mitosis, desde el final de profase hasta telofase.[64] Una fracción de esta proteína también se encuentra asociada con los microtúbulos y los polos del huso, y una parte aparece de forma difusa en el citoplasma.
- La destrucción de la ciclina B y la inactivación de CDK se inicia en los polos del huso. Clute y Pines midieron la inactivación de ciclina B1 en tiempo real, y vieron que ésta se inicia tan pronto como el último cromosoma se alinea en la placa metafásica, momento en que se inactiva el punto de control de la mitosis.[70]
Maquinaria del punto de control de la mitosis: proteínas implicadas
En Saccharomyces cerevisiae
Una gran parte de los componentes del checpunto de control de la mitosis se identificaron en dos análisis genéticos realizados en S. cerevisiae en 1991. Estos dos estudios eran similares, aunque presentaban diferencias sutiles: Hoyt y colaboradores[15] utilizaron cantidades mayores de benomyl (esta droga produce la despolimerización de los microtúbulos y se utilizó tres veces la cantidad necesaria para inhibir el crecimiento) que la utilizada por Li y Murray,[14] quienes utilizaron una cantidad que reduce, pero no bloquea el ensamblaje del huso. El grupo de Hoyt aisló cepas mutantes que morían en respuesta al tratamiento, ya que no se detenían en el ciclo celular, como era el caso de las cepas tipo salvaje. En el caso de Li y Murray, identificaron cepas mutantes que salían de mitosis antes de ensamblar un huso funcional y morían posteriormente. Las cepas mutantes de ambos estudios, denominadas “gemación desinhibida por benzimidazol” (budding uninhibited by benzimidazole, Bub1, Bub2, Bub3) las del primer estudio y “deficientes en detención de mitosis” (mitotic arrest deficient, Mad1, Mad2, Mad3) las del segundo, formaban microtúbulos normalmente y podían ensamblar un huso, lo cual indicaba que la sensibilidad al tratamiento no se debía a la presencia de un problema estructural, sino más bien a un defecto en un mecanismo de regulación.
En otros estudios se identificaron otras proteínas que forman parte de este mecanismo:
- Mps1p (monopolar spindle, “huso monopolar”). Esta es una proteína esencial requerida para la duplicación de los polos del huso en levaduras. La sobreexpresión de Mps1p afecta tanto a la integridad de los polos del huso como a la actividad del punto de control de la mitosis.[18][71]
- Cdc20 interacciona con los componentes del punto de control de la mitosis Mad1, Mad2 y Mad3 en levaduras.[72] El estudio realizado por Hwang y colaboradores en 1998 mostraba que formas mutantes de cdc20 que no pueden unirse a las proteínas Mad no consiguen activar el punto de control de la mitosis y detener la célula en metafase, lo que implica que Cdc20 es la diana del mecanismo de acción del punto de control.

Teniendo en cuenta los componentes mencionados anteriormente y las interacciones genéticas identificadas entre ellos, la transición de mitosis a interfase en S. cerevisiae se ha descrito como dos puntos de control interconectados:[73]
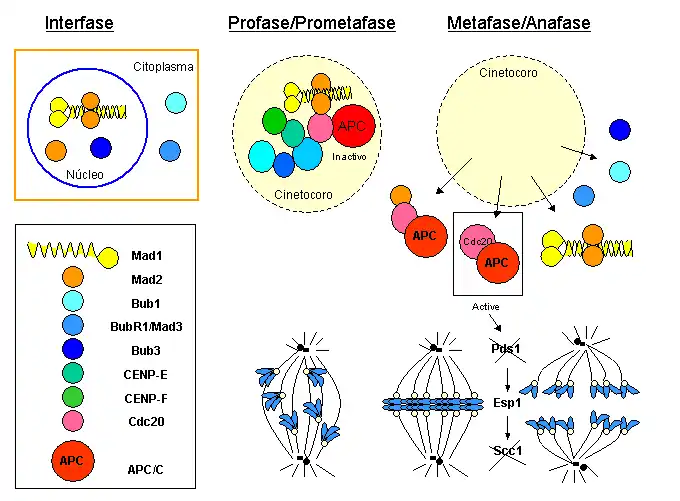
- La transición metafase-anafase, controlada por el punto de control de la mitosis: esta ruta converge en Mad1-Mad2 y Mad3, proteínas que se unen a Cdc20 e inhiben la actividad de APC/CCdc20, de forma que Pds1p/Securina permanece unida a Esp1/Separasa. Cuando todos los cromosomas se congregan en la placa metafásica, Mad2 y Mad3 se separan de Cdc20, con lo cual el APC/CCdc20 se activa, marcando Pdsp1/Securina para su degradación, de forma que Esp1/Separasa se libera y corta las cohesinas, de manera que las cromátidas hermanas se separan, iniciando así la anafase.
- La salida de mitosis controlada por la red de salida de mitosis (mitotic exit network o MEN), que marca Clb2p (ciclina B) para su degradación.
En vertebrados
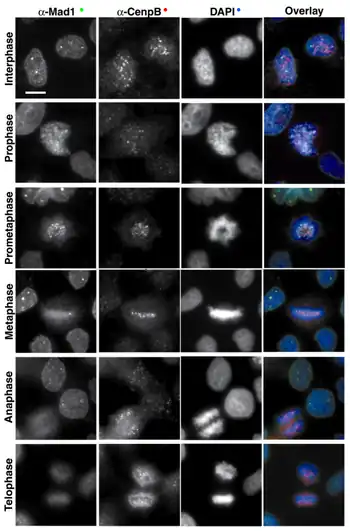
Se han identificado homólogos de las proteínas de S. cerevisiae del punto de control de la mitosis en todos los organismos estudiados, desde las levaduras de fisión hasta vertebrados: Schizosaccharomyces pombe,[74] Zea mays,[75] Caenorhabditis elegans,[76] Xenopus laevis,[77] Drosophila melanogaster,[78] Mus musculus[79] y Homo sapiens.[80]
La proteína que presenta una diferencia más llamativa entre S. cerevisiae y vertebrados es Mad3, que en vertebrados es una kinasa que por su similitud a Bub1 se denomina BubR1 o Bub1b.[80] Por otro lado, en vertebrados el punto de control de la mitosis es más complejo que en S. cerevisiae, probablemente debido a que la estructura mitótica presenta más componentes: mientras que en levaduras se ancla un único microtúbulo por cinetocoro, en vertebrados a un único cinetocoro se anclan alrededor de veinte microtúbulos.[81] En consonancia con esta mayor complejidad estructural, en vertebrados se han identificado componentes del punto de control de la mitosis que no tienen homología con ninguna proteína en levaduras; este es el caso de la proteína motora Cenp-E,[82] entre otras. Otra diferencia significativa es que mientras que en levaduras el punto de control de la mitosis se activa únicamente en respuesta a la detección de un fallo antes de la congregación cromosómica, en vertebrados el punto de control se activa de forma constitutiva al inicio de la mitosis, y permanece activado hasta el momento en que el último cromosoma alcanza la placa metafásica. En ese momento el punto de control se inactiva, y se dispara el inicio de la anafase con la destrucción de las cohesinas que mantenían unidas las cromátidas hermanas.
A diferencia de las proteínas centroméricas estructurales (como Cenp-B), que se mantienen en esta posición a lo largo de toda la mitosis, incluida telofase, los componentes de este punto de control presentan una localización característica en mitosis:[83] durante profase y prometafase se concentran en los cinetocoros de los cromosomas aún no anclados al huso, y esta localización se pierde en el momento que los cinetocoros capturan microtúbulos, de manera que en metafase, cuando todos los cromosomas están congregados en la placa metafásica, se liberan las proteínas reguladoras. La desaparición de las proteínas reguladoras de los cinetocoros marca el momento en que los cromosomas han alcanzado la placa y se encuentran en tensión bipolar. Es entonces cuando las proteínas reguladoras (Mad1-Mad2 y BubR1) que se unen e inhiben Cdc20, lo liberan, permitiendo la activación del APC/CCdc20, que dispara la separación de las cromátidas hermanas.
Como las alteraciones de las proteínas reguladoras de la mitosis pueden ser la causa de la formación de células aneuploides, y dado que éste es un suceso frecuente en el cáncer, inicialmente se supuso que estos genes podían estar mutados en los tejidos cancerosos.[84] Estudios posteriores de diferentes laboratorios no han detectado una mayor frecuencia de mutaciones en estos genes en cáncer, aunque el punto de control de la mitosis no funciona correctamente en muchos casos.[85] Sin embargo, lo que sí se ha identificado es que variaciones en los niveles fisiológicos de estas proteínas (como Mad2, por ejemplo) están asociadas con la generación de aneuploidía y tumorigénesis, lo que ha podido demostrarse utilizando modelos animales.[86]
Referencias
- Conlon I., Raff M. (1999). «Size control in animal development». Cell 96 (2). 235-244.
- Meraldi P., Lukas J., Fry A.M., Bartek J., Nigg E.A. (1999). «Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A». Nat.Cell Biol. 1 (2). 88-93.
- Mayor, T. Meraldi, P., Stierhof, Y.D., Nigg, E.A., Fry, A.M. (1999). «Protein kinases in control of the centrosome cycle». FEBS Lett. 452 (1-2). 92-95. (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).
- Nicklas, R.B. (1997). «How cells get the right chromosomes». Science 275 (5300). 632-7.
- Loncarek J, Kisurina-Evgenieva O, Vinogradova T, Hergert P, La Terra S, Kapoor TM, Khodjakov A. (2007). «The centromere geometry essential for keeping mitosis error free is controlled by spindle forces.». Nature 450 (7170). 745-9.
- Dewar H., Tanaka K., Nasmyth K., Tanaka T.U. (2004). «Tension between two kinetochores suffices for their bi-orientation on the mitotic spindle.». Nature 428 (6978). 93-97.
- Cimini D., Wan X., Hirel C.B., Salmon E.D. (2006). «Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors.». Curr. Biol. 16 (17). 1711-1718.
- Gautschi O., Heighway J., Mack P.C., Purnell P.R., Lara P.N.Jr., Gandara D.R. (2008). «Aurora kinases as anticancer drug targets.». Clin. Cancer Res. 14 (6). 1639-1648.
- Schaar B.T., Chan G.K., Maddox P., Salmon E.D., Yen T.J. (1997). «CENP-E function at kinetochores is essential for chromosome alignment». J.Cell Biol. 139 (6). 1373-1382.
- Wood K.W., Sakowicz R., Goldstein L.S., Cleveland D.W. (1997). «CENP-E is a plus end-directed kinetochore motor required for metaphase chromosome alignment». Cell 91 (3). 357-366.
- Zirkle R.E. (1970). «Ultraviolet-microbeam irradiation of newt-cell cytoplasm: spindle destruction, false anaphase, and delay of true anaphase.». Radiat Res 41 (3). 516-537.
- Rieder C.L., Palazzo R.E. (1992). «Colcemid and the mitotic cycle». J. Cell Sci. 102 (Pt 3). 387-392.
- Burke D.J., Stukenberg P.T. (2008). «Linking Kinetochore-Microtubule Binding to the Spindle Checkpoint.». Developmental Cell 14 (4). 474-479.
- Li R., Murray A.W. (1991). «Feedback control of mitosis in budding yeast.». Cell 66 (3). 519-531.
- Hoyt M.A., Totis L., Roberts B.T. (1991). «S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function.». Cell 66 (3). 507-517.
- Neff M.W., Burke D.J. (1992). «A delay in the Saccharomyces cerevisiae cell cycle that is induced by a dicentric chromosome and dependent upon mitotic checkpoints.». Mol.Cell Biol. 12 (9). ISSN, PÁGINA/S. [3857-3864]
- Wells W.A., Murray A.W. (1996). «Aberrantly segregating centromeres activate the spindle assembly checkpoint in budding yeast.». J. Cell Biol. 133 (1). 75-84.
- Hardwick K.G., Weiss E., Luca F.C., Winey M., Murray A.W. (1996). «Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption.». Science 273 (5277). 953-956.
- Wang Y., Burke D.J. (1995). «Checkpoint genes required to delay cell division in response to nocodazole respond to impaired kinetochore function in the yeast Saccharomyces cerevisiae.». Mol. Cell Biol. 15 (12). 6838-6844.
- Spencer F., Hieter P. (1992). «Centromere DNA mutations induce a mitotic delay in Saccharomyces cerevisiae.». PNAS 89 (19). 8909-8912.
- Hardwick K.G., Li R., Mistrot C., Chen R.H., Dann P., Rudner A., Murray A.W. (1999). «Lesions in Many Different Spindle Components Activate the Spindle Checkpoint in the Budding Yeast Saccharomyces cerevisiae.». Genetics 152 (2). 509-518.
- McIntosh J.R. (1991). «Structural and mechanical control of mitotic progression.». Cold Spring Harb Symp Quant Biol 56. 613-619.
- Rieder C.L., Cole R.W., Khodjakov A., Sluder G. (1995). «The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores.». J. Cell Biol. 130 (4). 941-948.
- Li X., Nicklas R.B. (1997). «Tension-sensitive kinetochore phosphorylation and the chromosome distribution checkpoint in praying mantid spermatocytes.». J. Cell Sci. 110 (Pt 5). 537-545.
- Rieder C.L., Khodjakov A., Paliulis L.V., Fortier T.M., Cole R.W., Sluder G. (1997). «Mitosis in vertebrate somatic cells with two spindles: implications for the metaphase/anaphase transition checkpoint and cleavage.». PNAS 94 (10). 5107-5112.
- Hirano T. (2000). «Chromosome cohesion, condensation, and separation.». Annual Review of Biochemistry 69 (1). 115-144.
- Tanaka K., Hao Z., Kai M., Okayama H. (2001). «Establishment and maintenance of sister chromatid cohesion in fission yeast by a unique mechanism.». EMBO J. 20 (20). 5779-5790.
- Gonzalez C., Casal Jimenez J., Ripoll P., Sunkel C. E. (1991). «The spindle is required for the process of sister chromatid separation in Drosophila neuroblasts.». Exp Cell Res 192 (1). 10-15.
- Losada A., Hirano T. (2001). «Shaping the metaphase chromosome: coordination of cohesion and condensation.». Bioessays 23 (10). 924-35.
- Bernard, Pascal Maure J-F., Partridge J.F., Genier S., Javerzat J-P., Allshire R.C. (2001). «Requirement of Heterochromatin for Cohesion at Centromeres.». Science 294 (5551). 2539-2542.
- Nonaka N., Kitajima T., Yokobayashi S., Xiao G., Yamamoto M., Grewal S. I. S., Watanabe Y. (2002). «Recruitment of cohesin to heterochromatic regions by Swi6/HP1 in fission yeast.». Nat Cell Biol 4 (1). 89-93.
- Hall I.M., Noma K-I., Grewal, Shiv I.S. (2003). «RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast.». PNAS 100 (1). 193-198.
- Fukagawa T., Nogami M., Yoshikawa M., Ikeno M., Okazaki T., Takami Y., Nakayama T., Oshimura M. (2004). «Dicer is essential for formation of the heterochromatin structure in vertebrate cells.». Nat Cell Biol 6 (8). 784-791.
- Weber S.A., Gerton J.L., Polancic J.E., DeRisi J.L., Koshland D., Megee P.C. (2004). «The Kinetochore Is an Enhancer of Pericentric Cohesin Binding.». PLoS Biology 2 (9). e260.
- Prasanth, S.G.; Prasanth, K.V.; Siddiqui, K.; Spector, D.L.; Stillman, B. (2004), «Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance», The EMBO Journal 23: 2651-2663, doi:10.1038/sj.emboj.7600255.
- Shimada, K.; Gasser, S.M. (2007), «The Origin Recognition Complex Functions in Sister-Chromatid Cohesion in Saccharomyces cerevisiae», Cell 128 (1): 85-99, doi:10.1016/j.cell.2006.11.045.
- Kato, H.; Matsunaga, F.; Miyazaki, S.; Yin, L.; D'urso, G.; Tanaka, K.; Murakami, Y. (2008), «Schizosaccharomyces pombe Orc5 plays multiple roles in the maintenance of genome stability …», Cell cycle (Georgetown, Tex.) 7 (8).
- Hauf S., Watanabe Y. (2004). «Kinetochore orientation in mitosis and meiosis.». Cell 119 (3). 317-327.
- Sonoda E., Matsusaka T., Morrison C., Vagnarelli P., Hoshi O., Ushiki T., Nojima K., Fukagawa T., Waizenegger I.C., Peters J.M., Earnshaw W.C., Takeda S. (2001). «Scc1/Rad21/Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells.». Dev Cell 1 (6). 759-770.
- Vass S., Cotterill S., Valdeolmillos A.M., Barbero J.L., Lin E., Warren W.D., Heck M.M. (2003). «Depletion of Drad21/Scc1 in Drosophila cells leads to instability of the cohesin complex and disruption of mitotic progression.». Curr Biol 13 (3). 208-218.
- Haering C.H., Lowe J., Hochwagen A., Nasmyth K. (2002). «Molecular architecture of SMC proteins and the yeast cohesin complex.». Mol Cell 9 (4). 773-788.
- Hirano T. (1999). «SMC-mediated chromosome mechanics: a conserved scheme from bacteria to vertebrates?». Genes Dev. 13 (1). 11-19.
- Hirano T. (2000). «Chromosome cohesion, condensation, and separation.». Annual Review of Biochemistry 69 (1). 115-144.
- Ciosk R., Zachariae W., Michaelis C., Shevchenko A., Mann M., Nasmyth K. (1998). «An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast.». Cell 93 (6). ISSN, 1067-1076.
- Alexandru G., Uhlmann F., Mechtler K., Poupart M., Nasmyth K. (2001). «Phosphorylation of the cohesin subunit scc1 by polo/cdc5 kinase regulates sister chromatid separation in yeast.». Cell 105 (4). 459-472.
- Leismann O., Herzig A., Heidmann S., Lehner C.F. (2000). «Degradation of Drosophila PIM regulates sister chromatid separation during mitosis.». Genes Dev. 14 (17). 2192-2205.
- Zur A., Brandeis M. (2001). «Securin degradation is mediated by fzy and fzr, and is required for complete chromatid separation but not for cytokinesis.». EMBO J. 20 (4). 792-801.
- Sumara I., Vorlaufer E., Gieffers C., Peters B.H., Peters, J.-M. (2000). «Characterization of Vertebrate Cohesin Complexes and Their Regulation in Prophase.». J. Cell Biol. 151 (4). 749-762.
- Losada A., Yokochi T., Kobayashi R., Hirano T. (2000). «Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes.». J. Cell Biol. 150 (3). 405-416.
- Gimenez-Abian J.F., Sumara I., Hirota T., Hauf S., Gerlich D., de la Torre C., Ellenberg J., Peters J.-M. (2004). «Regulation of Sister Chromatid Cohesion between Chromosome Arms.». Current Biol. 14 (13). 1187-1193.
- Paliulis L.V., Nicklas R.B. (2004). «Micromanipulation of chromosomes reveals that cohesion release during cell division is gradual and does not require tension.». Curr. Biol. 14 (23). 2124-2129.
- Nakajima M., Kumada K., Hatakeyama K., Noda T., Peters J.-M., Hirota T. (2007). «The complete removal of cohesin from chromosome arms depends on separase». J. Cell Sci. 120 (23). 4188-4196.
- McGuinness B.E., Hirota T., Kudo N.R., Peters J.-M., Nasmyth K. (2005). «Shugoshin prevents dissociation of cohesin from centromeres during mitosis in vertebrate cells.». PLoS Biol. 3 (3). e86.
- Salic A., Waters J.C., Mitchison T.J. (2004). «Vertebrate shugoshin links sister centromere cohesion and kinetochore microtubule stability in mitosis.». Cell 118 (5). 567-578.
- Nigg E.A. (2001). «Mitotic kinases as regulators of cell division and its checkpoints.». Nature Reviews Molecular Cell Biology 2 (Jan). 21-32.
- Ohi R., Gould K.L. (1999). «Regulating the onset of mitosis.». Curr Opin Cell Biol 11 (2). 267-273.
- Gould K.L., Moreno S., Tonks N.K., Nurse P. (1990). «Complementation of the mitotic activator, p80cdc25, by a human protein-tyrosine phosphatase.». Science 250 (4987). 1573-1576.
- Visintin R., Prinz S., Amon A. (1997). «CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis.». Science 278 (5337). 460-463.
- Schwab M., Lutum A.S., Seufert W. (1997). «Yeast Hct1 is a regulator of Clb2 cyclin proteolysis.». Cell 90 (4). 683-693.
- Cohen-Fix O., Peters J.-M., Kirschner M.W., Koshland D. (1996). «Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p.». Genes Dev. 10 (24). 3081-3093.
- Glotzer M., Murray A.W., Kirschner M.W. (1991). «Cyclin is degraded by the ubiquitin pathway.». Nature 349 (6305). 132-138.
- Murray A. (1995). «Cyclin ubiquitination: the destructive end of mitosis.». Cell 81 (2). 149-152.
- Peters J.-M. (1998). «SCF and APC: the Yin and Yang of cell cycle regulated protelysis.». Current Opinion in Cell Biology 10. 759-768.
- Kallio M., Weinstein J., Daum J.R., Burke D.J., Gorbsky G.J. (1998). «Mammalian p55CDC mediates association of the spindle checkpoint protein Mad2 with the cyclosome/anaphase-promoting complex, and is involved in regulating anaphase onset and late mitotic events.». J Cell Biol 14 (6). 1393-1406.
- Lahav-Baratz S., Sudakin V., Ruderman J.V., Hershko A. (1995). «Reversible phosphorylation controls the activity of cyclosome-associated cyclin-ubiquitin ligase.». PNAS 92 (20). 9303-9307.
- Zachariae W., Schwab M., Nasmyth K., Seufert W. (1998). «Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex.». Science 282 (5394). 1721-1724.
- Cohen-Fix O., Koshland D. (1999). «Pds1p of budding yeast has dual roles: inhibition of anaphase initiation and regulation of mitotic exit.». Genes Dev. 13 (15). 1950-1959.
- Kramer E.R., Scheuringer N., Podtelejnikov A.V., Mann M., Peters J.-M. (2000). «Mitotic regulation of the APC activator proteins CDC20 and CDH1.». Mol. Biol. Cell 11 (5). 1555-1569.
- Tugendreich S., Tomkiel J., Earnshaw W., Hieter P. (1995). «CDC27Hs colocalizes with CDC16Hs to the centrosome and mitotic spindle and is essential for the metaphase to anaphase transition.». Cell 81 (2). 261-268.
- Clute P., Pines J. (1999). «Temporal and spatial control of cyclin B1 destruction in metaphase». Nat. Cell Biol. 1 (2). 82-87.
- Weiss E., Winey M. (1996). «The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint.». J. Cell Biol. 132 (1-2). 111-123.
- Hwang L.H., Lau L.F., Smith D.L., Mistrot C.A., Hardwick K.G., Hwang E.S., Amon A., Murray A.W. (1998). «Budding yeast Cdc20: a target of the spindle checkpoint.». Science 279 (5353). 1041-1044.
- Gardner R.D.,Burke D.J. (2000). «The spindle checkpoint: two transitions, two pathways.». Trends Cell Biol. 10 (4). 154-158.
- He X., Jones M.H., Winey M., Sazer S. (1998). «mph1, a member of the Mps1-like family of dual specificity protein kinases, is required for the spindle checkpoint in S. pombe.». J Cell Sci 111 (Pt 12). 1635-1647.
- Yu H.G., Muszynski M.G., Kelly Dawe R. (1999). «The Maize Homologue of the Cell Cycle Checkpoint Protein MAD2 Reveals Kinetochore Substructure and Contrasting Mitotic and Meiotic Localization Patterns.». J Cell Biol 145 (3). 425-435.
- Kitagawa R., Rose A.M. (1999). «Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans.». Nat.Cell Biol. 1 (8). 514-521.
- Chen R.H., Shevchenko A., Mann M., Murray A.W. (1998). «Spindle Checkpoint Protein Xmad1 Recruits Xmad2 to Unattached Kinetochores.». J Cell Biol 143 (2). 283-295.
- Basu J., Bousbaa H., Logarinho E., Li Z., Williams B.C., Lopes C., Sunkel C.E., Goldberg M.L (1999). «Mutations in the essential spindle checkpoint gene bub1 cause chromosome missegregation and fail to block apoptosis in Drosophila.». J Cell Biol 146 (1). 13-28.
- Taylor S.S., McKeon F. (1997). «Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage.». Cell 89 (5). 727-735.
- Taylor S.S., Ha E., McKeon F. (1998). «The human homologue of bub3 is required for kinetochore localization of bub1 and a Mad3/Bub1-related protein kinase.». J Cell Biol 142 (1). 1-11.
- McEwenB.F., Heagle A.B., Cassels G.O., Buttle K.F., Rieder C.L. (1997). «Kinetochore fiber maturation in PtK1 cells and its implications for the mechanisms of chromosome congression and anaphase onset.». J. Cell Biol. 137 (7). 1567-1580.
- YaoX.B., Abrieu A., Zheng Y., Sullivan K.F., Cleveland D.W. (2000). «CENP-E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint.». Nature Cell Biology 2 (8). 484-491.
- Martin-Lluesma S., Stucke V. M., Nigg E. A. (2002). «Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2.». Science 297 (5590). 2267-2270.
- Cahill D.P., Lengauer C., Yu J., Riggins G.J., Willson J.K., Markowitz S.D., Kinzler K.W., Vogelstein B. (1998). «Mutations of mitotic checkpoint genes in human cancers». Nature 392 (6673). 300-303.
- Haruki N., Saito H., Harano T., Nomoto S., Takahashi T., Osada H., Fujii Y., Takahashi T. (2001). «Molecular analysis of the mitotic checkpoint genes BUB1, BUBR1 and BUB3 in human lung cancers». Cancer Lett. 162 (2). 201-205.
- Sotillo R., Hernando E., Diaz-Rodriguez E., Teruya-Feldstein J., Cordon-Cardo C., Lowe S. W., Benezra R. (2007). «Mad2 overexpression promotes aneuploidy and tumorigenesis in mice.». Cancer Cell 11 (1). 9-23.
Enlaces externos
- Laboratorio de Ted Salmon: películas de células en división.
- Laboratorio de Andrea Musacchio: esquemas del checkpoint de mitosis.
 Datos: Q9064829
Datos: Q9064829