Reacción SN1
La reacción SN1 es una reacción de sustitución en química orgánica. "SN" indica que es una sustitución nucleofílica y el "1" representa el hecho de que la etapa limitante es unimolecular.[1][2] La reacción involucra un intermediario carbocatión y es observada comúnmente en reacciones de halogenuros de alquilo secundarios o terciarios, o bajo condiciones fuertemente acídicas, con alcoholes secundarios y terciarios. Con los halogenuros de alquilo primarios, sucede la reacción SN2, alternativa. Entre los químicos inorgánicos, la reacción SN1 es conocida frecuentemente como el mecanismo disociativo. Un mecanismo de reacción fue propuesto por primera vez por Christopher Ingold y colaboradores en 1940.[3]
Mecanismo
Un ejemplo de una reacción que tiene lugar con un mecanismo de reacción SN1 es la hidrólisis del bromuro de tert-butilo con agua, formando alcohol tert-butílico:
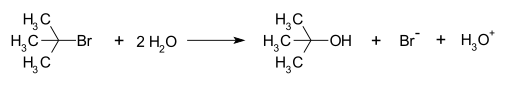
Esta reacción SN1 tiene lugar en tres etapas:
- Formación de un carbocatión de tert-butilo, por la separación de un grupo saliente (un anión bromuro) del átomo de carbono: esta etapa es lenta y es reversible.[4]
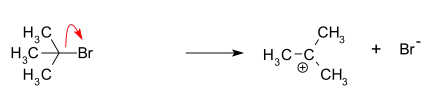
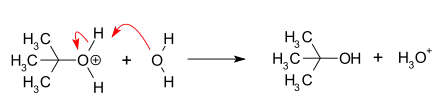
- Ataque nucleofílico: el carbocatión reacciona con el nucleófilo. Si el nucleófilo es una molécula neutra (por ejemplo, un disolvente), se requiere un tercer paso para completar la reacción. Cuando el disolvente es agua, el intermediario es un ion oxonio. Esta etapa de reacción es rápida.

- Desprotonación: La eliminación de un protón en el nucleófilo protonado por el agua actuando como base conduce a la formación del alcohol y un ion hidronio. Esta etapa de la reacción es rápida.
Cinética
En contraste con las reacciones SN2, las reacciones SN1 suceden en dos etapas (excluyendo cualquier protonación o deprotonación). La etapa limitante es la primera etapa, así que la velocidad global de la reacción es esencialmente igual a la formación del carbocatión y no involucra al nucleófilo atacante. En consecuencia, la nucleofilicidad es irrelevante, y la velocidad de reacción global depende sólo de la concentración del sustrato.
- velocidad = k[sustrato]
En 1954 se encontró que la adición de una cantidad pequeña de perclorato de litio a ciertas reacciones de acetólisis (por ejemplo del tosilato del colesterol) conducían a un aumento significativo de la velocidad de reacción.[5] A partir de este efecto de la sal especial el mecanismo general fue refinado para incluir un par iónico de contacto con el catión y el anión juntos en una jaula de disolvente, que luego se disocia en un par iónico separado por disolvente, y luego en iones libres. Todas las interconversiones son reversibles y la sal agregada evita la reformación del par de contacto a partir del par separado por disolvente.
En algunos casos, la reacción SN1 sucederá a una velocidad anormalmente alta, debido a la participación de grupos vecinales. Los grupos vecinales frecuentemente abaten la barrera energética requerida para la formación del intermediario carbocatiónico.
Alcance de la reacción
El mecanismo SN1 tiende a dominar cuando el átomo de carbono central está rodeado por grupos voluminosos, debido a que tales grupos inhiben estéricamente la reacción SN2. Además, los sustituyentes voluminosos en el carbono central aumentan la velocidad de formación del carbocatión porque libera la tensión estérica cuando ocurre. El carbocatión resultante también está estabilizado tanto por estabilización inductiva y por hiperconjugación de los grupos alquilo adyacentes. El postulado de Hammond-Leffler sugiere que esto también incrementará la velocidad de la formación del carbocatión. De ahí que el mecanismo SN1 domina en las reacciones en centros de alquilo terciario, y es observada en centros de alquilo secundario en presencia de nucleófilos débiles.
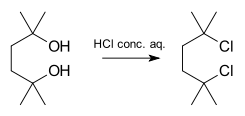
Un ejemplo de una reacción que procede en un mecanismo SN1 es la síntesis del 2,5-dicloro-2,5-dimetilhexano a partir del diol correspondiente con ácido clorhídrico concentrado:[6]
Estereoquímica
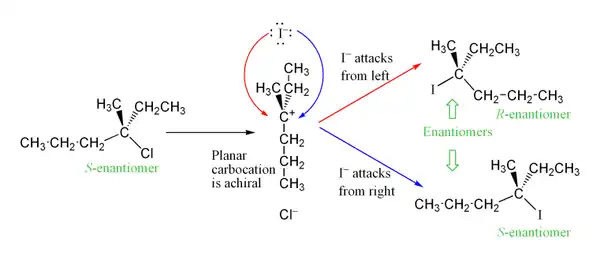
Debido a que el carbocatión intermediario es plano, el carbono central no es un estereocentro. Incluso si hubiera sido un estereocentro antes de ser un carbocatión, la configuración original en dicho átomo se pierde. En vez de ello, el carbono central puede ser proquiral. El ataque nucleofílico puede ocurrir desde cualquier lado del plano, así que el producto puede consistir de una mezcla de dos estereoisómeros. En efecto, si el átomo de carbono central es el único estereocentro en la reacción, puede producirse una racemización. Esto contrasta con los mecanismos SN2, donde la configuración quiral del sustrato se invierte. Sin embargo, usualmente se ve un exceso de inversión, puesto que el grupo saliente puede permanecer en la proximidad del intermediario carbocatiónico por un corto tiempo, y bloquear el ataque nucleofílico. Por ejemplo, en la reacción del S-3-cloro-3-metilhexano con anión yoduro, el intermediario carbocatiónico está libre del grupo saliente y es, en consecuencia, aquiral, y habría una oportunidad idéntica de ataque por cualquier lado. Esto conduce a una mezcla de R-3-yodo-3-metilhexano y S-3-yodo-3-metilhexano:
Reacciones laterales
Las dos reacciones comunes son la reacción de eliminación y el rearreglo de carbocatión. Si la reacción es llevada a cabo bajo condiciones de calor (que favorecen un incremento en la entropía), conduciendo a la formación de un alqueno. Incluso si la reacción se lleva a cabo en frío, se puede formar algo de alqueno. Si se intenta llevar a cabo una reacción SN1 usando un nucleófilo fuertemente básico, tal como un ion hidróxido o metóxido, se formará nuevamente el alqueno, esta vez por un mecanismo de eliminación E2. Esto será verdad especialmente cuando la reacción es calentada. Finalmente, si el intermediario carbocatiónico puede rearreglarese a un carbocatión más etable, dará un producto derivado del carbocatión más estable, en vez del producto de sustitución simple.
Efectos del solvente
Dado que la reacción SN1 involucra la formación de un intermediario carbocatiónico inestable en la etapa limitante, cualquier cosa que facilite esto acelerará la reacción. Los solventes de elección son tanto de naturaleza polar (para estabilizar los intermediarios iónicos en general) y prótica (para solvatar el grupo saliente en particular). Los solventes próticos polares incluyen al agua y alcoholes, que también actúan como nucleófilos.
Véase también
- Sustitución nucleofílica acílica
- Participación del grupo vecinal
Referencias
- L. G. Wade, Jr., Organic Chemistry, 6th ed., Pearson/Prentice Hall, Upper Saddle River, New Jersey, USA, 2005
- J. March, Advanced Organic Chemistry, 4th ed., Wiley, New York, 1992.
- Leslie C. Bateman, Mervyn G. Church, Edward D. Hughes, Christopher K. Ingold and Nazeer Ahmed Taher (1940). «188. Mechanism of substitution at a saturated carbon atom. Part XXIII. A kinetic demonstration of the unimolecular solvolysis of alkyl halides. (Section E) a general discussion». Journal of the Chemical Society (Resumed): 979. doi:10.1039/JR9400000979.
- Peters, K. S. (2007). «Nature of Dynamic Processes Associated with the SN1 Reaction Mechanism». Chem. Rev. 107 (3): 859-873. doi:10.1021/cr068021k.
- S. Winstein, E. Clippinger, A. H. Fainberg, and G. C. Robinson (1954). «Salt effects and ion-pairs in solvolysis». J. Am. Chem. Soc. 76: 2597. doi:10.1021/ja01638a093.
- Synthesis of 2,5-Dichloro-2,5-dimethylhexane by an SN1 Reaction Carl E. Wagner and Pamela A. Marshall , J. Chem. Educ., 2010, 87 (1), pp 81–83 doi 10.1021/ed8000057
Lecturas adicionales
- Electrophilic Bimolecular Substitution as an Alternative to Nucleophilic Monomolecular Substitution in Inorganic and Organic Chemistry / N.S.Imyanitov. J. Gen. Chem. USSR (Engl. Transl.) 1990; 60 (3); 417-419.
- Unimolecular Nucleophilic Substitution does not Exist! / N.S.Imyanitov. SciTecLibrary
Enlaces externos
| Control de autoridades |
|
|---|
 Datos: Q838130
Datos: Q838130 Multimedia: SN1 reactions / Q838130
Multimedia: SN1 reactions / Q838130