Síntesis de oligonucleótidos
La síntesis de oligonucleótidos es la síntesis química de fragmentos relativamente cortos de ácido nucleico con una secuencia estructural química definida. La técnica es extremadamente útil en la práctica actual en laboratorios, porque provee un acceso rápido y barato a oligonucleótidos hechos a la medida con una secuencia deseada. Mientras que las enzimas sintetizan ADN y ARN en una dirección de 5' a 3', la síntesis de oligonucleótidos se lleva a cabo en el sentido opuesto, en la dirección 3' a 5'. Actualmente, el proceso está implementado como una síntesis en fase sólida usando el método de la fosforoamidita y bloques de construcción de fosforoamidita derivados de 2'-deoxinucleósidos (dA, dC, dG y T), ribonucleósidos (A, C, G y U), o nucleósidos químicamente modificados, como los ANB.
Para obtener el oligonucleótido deseado, se acoplan secuencialmente los bloques de construcción a la cadena de oligonucleótidos en el orden requerido por la secuencia del producto (ver Ciclo de Síntesis más abajo). El proceso ha sido automatizado completamente desde finales de la década de 1970. Al completarse el ensamblado de la cadena, se libera el producto de la fase sólida a la solución, luego se le desprotege, y finalmente se le recolecta. La existencia de reacciones secundarias establece límites prácticos a la longitud de los oligonucleótidos sintéticos (hasta aproximadamente 200 residuos de nucleótido) debido a que el número de errores se acumulan con la longitud del oligonucleótido sintetizado.[1] Los productos suelen ser aislados mediante HPLC para obtener los oligonucleótidos deseados en una alta pureza. Típicamente, los oligonucleótidos sintéticos son moléculas de DNA o RNA de una sola hebra de aproximadamente 15–25 bases de longitud.
Los oligonucleótidos tienen muchas aplicaciones tanto en el campo de biología molecular como en medicina. Son comúnmente utilizados como oligonucleótidos de antisentido, SiRNA, partidores para secuenciación de ADN y amplificación, sonda genética para detectar ADN o ARN complementario vía hibridación molecular, herramientas para la introducción dirigida de mutaciones y sitios de restricción, y para la síntesis artificial de genes.
Historia
La evolución de la síntesis de oligonucleótidos ha visto cuatro métodos principales de formación de los enlaces internucleosídicos y ha sido revisada en la literatura con gran detalle.[2][3][4]
Primeros trabajos y síntesis contemporánea de H-fosfonato
A principios de la década de 1950, el grupo de Alexander Robert Todd fue pionero en los métodos de H-fosfonato y triéster de fosfato para la síntesis de oligonucleótidos.[5][6] La reacción de los compuestos 1 y 2 para formar el diéster de H-fosfonato 3 es una reacción de copulación en solución, mientras que la de los compuestos 4 y 5 para dar 6 es una copulación fosfotriéster (ver síntesis fosfotriéster más abajo).
Treinta años más tarde, este trabajo inspiró, independientemente, a dos grupos de investigación para adoptar la química del H-fosfonato a la síntesis de fase sólida, usando monoésteres H-fosfonato de nucleósidos 7 como bloques de construcción, y cloruro de pivaloílo, cloruro de 2,4,6-triisopropilsulfonilo (TPS-Cl), y otros compuestos como activadores.[7][8] La implementación práctica del método del H-fosfonato resultó en un ciclo sintético muy corto y simple, consistiendo de solo dos etapas, detritilación y copulación (Esquema 2). La oxidación de los enlaces diéster H-fosfonato internucleosídicos en 8 a enlaces fosfodiéster en 9 (X = O) con una solución de yodo en piridina acuosa es llevada a cabo al final del ensamblado de la cadena, como un paso en el ciclo sintético. Alternativamente, 8 puede ser convertido en el fosforotioato 10 [9][10][11][12] o en fosforoenolato 11 (X= Se),[13] o oxidado por CCl4 en presencia de aminas primarias o secundarias a análogos de fosforamidato 12.[14][15] El método es muy conveniente en varios tipos de modificaciones de fosfato(fosfato/fosforotioato/fosforamidato) y puede ser introducido al mismo oligonucleótido para la modulación de sus propiedades.[16][17][18]
Muy a menudo, la construcción de bloques de H-fosfonato está protegida en el grupo 5'-hidroxi y en el grupo amino de las bases nitrogenadas Adenina, Citosina y Guanina de la misma manera que los bloques de construcción de la fosforamidita (ver abajo). Sin embargo, la protección del grupo amino no es obligatoria.[19][20]


Síntesis de fosfodiéster
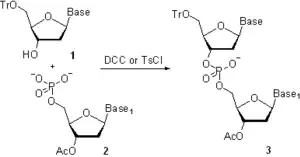
En la década de 1950, Har Gobind Khorana y sus colaboradores desarrollaron un método de fosfodiéster donde el 3’-O-acetilnucleósido-5’-O-fosfato 2(Esquema 3) se activaba con N,N’-diciclohexilcarbodiimida (DCC) o cloruro de 4-toluensulfonilo (Ts-Cl) y un nucleósido 5’-O protegido 1 se hizo reaccionar con la especie activada para dar un monofosfato de dinucleósido protegido 3.[21] Tras la eliminación del grupo acetilo 3’-O usando hidrólisis alcalina, se llevó a cabo una posterior elongación de la cadena. Siguiendo esta metodología, se sintetizaron conjuntos de tri- y tetradeoxirribonucleótidos, los que fueron convertidos enzimáticamente en oligonucleótidos más largos, lo que permitió la elucidación del código genético. La principal limitación del método del fosfodiéster consistía en la formación de oligómeros de pirofosfato y oligonucleótidos ramificados en el fosfato internucleosídico. El método parece ser un paso atrás desde la química más selectiva descrita anteriormente; sin embargo, en aquel entonces, no habían sido introducidos la mayor parte de grupos protectores de fosfato que hay hoy en día. La falta de una estrategia de protección conveniente necesitaba tomar una retirada a una química más lenta y menos selectiva para lograr el objetivo último del estudio.[2]
Síntesis de fosfotriéster
En la década de 1960, grupos dirigidos por R. Letsinger[22] y C. Reese[23] desarrollaron una solución basada en fosfotriéster. La diferencia que define de la solución basada en fosfodiéster era la protección de la entidad fosfato en el bloque constructor 1 (Esquema 4) y en el producto 3 con el grupo 2-cianoetilo. Esto precluía la formación de oligonucleótidos ramificados en el fosfato internucleosídico. La mayor selectividad del método permitió el uso de agentes copulantes y catalizadores más eficientes,[24][25] lo que redujo dramáticamente la duración de la síntesis. El método, inicialmente desarrollado para la síntesis en fase de solución, fue también implementado en poliestireno,[26] lo que inició un esfuerzo de investigación masivo en síntesis en fase sólida de oligonucleótidos y condujo eventualmente a la automatización del ensamblaje en las cadenas de oligonucleótidos.

Síntesis de triéster fosfito
En la década de 1970, sustancialmente más reactivos P(III) derivados de nucleósidos, 3'-O-clorofosfito, fueron usadas de manera exitosa en la formación de enlaces internucleosídicoss.[27] . Esto llevó al descubrimiento de la metodología del triéster fosfito. El grupo liderado por M. Caruthers tuvo la ventaja de tener 1H-tetrasolidofosfitos menos agresivos y más selectivos y por lo tanto implementaron el método en una fase sólida.[28] Poco tiempo después, los trabajadores del mismo grupo mejoraron el método usando nucleósidos fosforamiditos más estables como bloques de construcción.[29] El usar 2-cianoetil como grupo protector del fosfito.[30] en lugar de un grupo metilo.[31][32] más difícil de usar llevó a los nucleósidos fosforamiditos que se usan actualmente en la síntesis de oligonucleótidos (ver los bloques de construcción de fosforamiditos). Después vinieron muchas mejoras en la fabricación de los bloques de construcción, en los sintetizadores de oligonucleótidos y en los protocolos de sintetización que hicieron que la química de fosforamiditos fuera un método muy seguro y fácil para la preparación de oligonucleótidos sintéticos.[33]
Síntesis por medio del método de fosforamiditos
Fosforamiditos de nucleósidos
Como fue mencionado previamente, los nucleótidos de origen natural (nucleósido-3' o fosfatos 5') y sus análogos de fosfodiéster no son lo suficientemente reactivos para poder llevar a cabo la aceleración de la preparación sintética de oligonucleótidos en altos rendimientos.
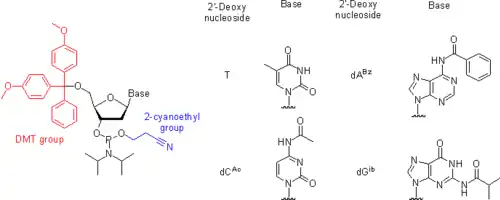
La selectividad y el tipo de formación de los enlaces internucleosídicos aumenta drásticamente al usar 3'-O-(N,N-diisopropil fosforamidita) derivados de nucleósidos (fosforamiditos de nucleósidos) que tienen una función de bloques de construcción en el método de triéster fosfito. Para prevenir una reacción secundaria no deseada, todos los demás grupos funcionales presentes en los nucleósidos tienen que ser no reactivos (protegidos) uniendo grupos protectores. Después de que el ensamblado de la cadena de oligonucleótidos ha sido completada, todos los grupos protectores son removidos para dar lugar a los oligonucleótidos deseados. A continuación se muestran los grupos protectores usados actualmente que se encuentran disponibles comercialmente[34][35][36][37] y los fosforamiditos de nucleósidos más comunes en los bloques de construcción:
- El grupo 5'-hidroxilo está protegido por un grupo ácido-lábil DMT (4,4'-dimetoxitritil).
- Timina y uracilo, bases nitrogenadas de timidina y uridina, respectivamente, no tienen grupos amino exocíclicos y por lo tanto no requieren ninguna protección.
- La guanosina y la 2'-deoxiguanosina tienen un grupo amino exocíclico, su basicidad es baja al grado en el que no reacciona con fosforamiditos bajo las condiciones de una reacción de copulación. Sin embargo, un fosforamidito derivado de N2-no protegido 5'-O-DMT-2'-desoxiguanosina es poco soluble en acetonitrilo, el solvente que es comúnmente usado en la síntesis de oligonucleótidos.[38] En contraste, las versiones de N2-protegidas del mismo compuesto son solubles en acetonitrilo y por lo tanto son muy usadas. Las bases nitrogenadas adenina y citosina tienen grupos amino exocíclicos reactivos con los fosforamiditos activados bajo las condiciones de una reacción de copulación. Bajo el uso de pasos adicionales en el ciclo de sintetización[39][40] o agentes alternativos de copulación y sistemas de solvatación,[38] el ensamblaje de la cadena de oligonucleótidos se puede llevar a cabo usando dA y dC fosforamiditos con grupos amino no protegidos. Sin embargo, estos intentos se encuentran actualmente en el campo de investigación. La síntesis de oligonucleótidos, los grupos amino exocíclicos en los nucleósidos son protegidos permanentemente en todo el proceso de ensamblaje de la cadena de oligonucleótidos.
La protección de los grupos amino exocíclicos tiene que ser ortogonal al grupo 5'-hidroxilo porque el último es removido al final de cada ciclo sintético. La estrategia más fácil de implementar y por lo tanto la más aceptada es en la que los grupos aminos exocíclicos tienen una protección base-lábil. Lo más común es usar dos esquemas de protección.
- En la primera, el esquema estándar y más robusto (Figura), protección de Bz (benzonil) es usada para A, dA y dC, mientras que G y dG son protegidas por un grupo isobutritil. Recientemente, el grupo Ac (acetilo) ha sido usado para proteger C y dC como se muestra en la figura.[41]
- En la segunda, en el esquema leve de protección, A y dA son protegidos con grupos de isobutritil[42] o fenoxiacetil (PAC).[43] C y dC tienen protección de acetil,[41] y G y dG son protegidos con grupos de 4-isopropilfenoxiacetil (Pr-PAC)[44] o dimetilformamidino (dmf).[45] Los grupos de protección leve son removidos más fácilmente que los grupos de protección estándar. Sin embargo, los fosforamiditos que contienen estos grupos son menos solubles cuando están almacenados en solución.
- El grupos fosfito es protegido por un grupo de base-lábil 2-cianoétil.[32] Una vez que un fosforamidito ha sido unido al soporte sólido de oligonucleótidos y los restos del fosfato se han convertido en especies P(V), la presencia de la protección del fosfato no es necesaria para que las reacciones de copulación se lleven a cabo de manera exitosa.[46]
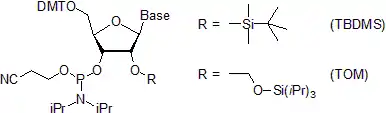
- En la síntesis de RNA, el grupo 2'-hidroxilo es protegido por el grupoTBDMS (t-butildimetilsilil)[47][48][49][50] o con el grupo TOM (tri-iso-propilsililoximetil),[51][52] los dos pueden ser removidos con un tratamiento del ion floruro.
- Los restos del fosfito también tienen al diisopropil amino (Pr2N) reactivo bajo condiciones ácidas. Cuando se activa, el grupo diisopropilamino se va para ser sustituido por el grupo 5'-hidroxilo que está unido al soporte del oligonucleótido (ver "paso 2: unión" en la parte de abajo)
Nucleósidos no fosforamiditos

Los nucleósidos no fosforamiditos son los reactivos de fosforamiditos que están diseñados para introducir diversas funciones en los extremos de los oligonucleótidos sintéticos o entre los residuos de los nucleótidos a la mitad de una secuencia. Para poder ser introducidos en una secuencia, un modificador no nucleosídico tiene que tener por lo menos 2 grupos hidroxilos, uno de ellos suele estar protegido por el grupo DMT, mientras que el otro contiene los restos del fosforamidito reactivo.
Los fosforamitos no nucleosídicos son usados para introducir los grupos deseados que no están disponibles en los nucleósidos naturales o que pueden ser introducidos de manera más fácil usando diseños químicos más simples. En el esquema para la demostración de la diversidad estructural y funcional disponible se puede ver una pequeña selección de reactivos de fosformaidito comercial. Estos reactivos sirven para la unión del fosfato 5'-terminal (1),[53] NH2 (2),[54] SH (3),[55] aldehído (4),[56] y grupos carboxílicos (5),[57] CC triples enlaces(6),[58] marcadores no radioactivos y desactivadores (ejemplificados por 6-FAM amidita 7[59] para la fijación de fluoresceína y DABCYL amidita 8,[60] respectivamente), modificadores hidrofílicos e hidrofóbicos (ejemplificados por hexaetilenglicol amidita 9[61][62] y colesterol amidita 10,[63] respectivamente) y amidita biotina 11.[64]
Ciclo sintético
La síntesis de oligonucleótidos se lleva a cabo por una adición gradual de residuos de nucleótidos al extremo 5' de la cadena creciente hasta que la secuencia deseada sea ensamblada. Cada adición tiene que ver con un ciclo sintético (esquema 5) y consiste en cuatro reacciones químicas:

Paso 1: El desbloqueo (detritilación)
El grupo DMT es removido con la solución de un ácido, puede ser 2% de ácido tricloroacético (TCA) o 3 % de ácido dicloroacético (DCA), en una solución inerte (diclorometano o tolueno). El catión DMT de color naranja es desteñido; el resultado de este paso es un oligonucleótido sólido unido al soporte que contiene una terminal 5'-hidroxilo libre. Es importante recordar que llevar a cabo una detritilación por un largo periodo de tiempo o con soluciones de ácidos más fuertes de las recomendadas puede llevar a una depurinación del soporte sólido ligado al oligonucleótido y por lo tanto reduce el rendimiento del producto deseado.
Paso 2: Unión
Una solución de 0.02-0.2 M de fosforamidito de nucleósido ( o una mezcla de varios fosforamiditos) en acetonitrilo es activado por una solución 0.2-0.7 M de un catalizador azol ácido, 1H-tetrazol, 5-ethylthio-1H-tetrazole,[65] 2-benzylthiotetrazole,[66][67] 4,5-dicyano imidazol,[68] o otros compuestos similares. En un análisis reciente, se puede encontrar mayor información en el uso de varios agentes de unión en la síntesis de oligonucleótidos.[69] La mezcla suele ser muy corta y ocurre en las líneas de fluido de los sintetizadores de oligonucleótidos (ver abajo) mientras que los componentes son entregados a los reactores que contienen el soporte sólido. La fosforamidita activada en 1.5-20 veces excede el soporte unido al material y es conectado con el soporte de sólido del principio (primera unión) o con un soporte unido al oligonucleótido precursor (siguientes uniones) cuyo grupo 5'-hidroxilo reacciona con los restos del fosforamidito activado del nucleósdio del fosforamidito que entra para formar un enlace fosfito triéster. La unión de los fosforamiditos 2'-desoxinucleosido es muy rápida y requiere, a pequeña escala, aproximadamente 20 s para poder llevarse a cabo. En contraste, 2'-O protegido de fosforamiditos de ribonucleósido requieren de 5 a 15 minutos para poder ser unidos en sistemas más complejos.[48][70][71][72] La reacción es muy sensible a la presencia de agua, especialmente cuando se usan soluciones diluidas de fosforamiditos, y normalmente se lleva a cabo por acetonitrilo anhídrido. Generalmente, a gran escala, en la síntesis, el exceso es menor y la concentración de fosforamiditos es mayor. La concentración del activador es determinada por su solubilidad en acetonitrilo, independientemente de la escala de la síntesis. Después de que la unión es completada, todos los reactivos que no fueron unidos y los productos no deseados se remueven mediante un lavado.
Paso 3: taponado
El paso del taponado se lleva a cabo mediante el tratamiento del soporte sólido unido al material con una mezcla de anhídrido acético y con 1-metilimidazol, o, menos común, DMAP como catalizadores en el método de fosforamiditos. Tiene dos propósitos:
- Después de completar la reacción de unión, un porcentaje pequeño del soporte de sólido unido a grupos 5'-OH (0.1 a 1%) no reacciona y por lo tanto necesita ser bloqueado permanentemente de la elongación de la cadena para prevenir la formación de oligonucleótidos con una deleción de bases internas, que se refiere normalmente como (n-1) cortameros. Los grupos 5'-hidroxilos son acetilados por una mezcla de taponado.
- También ha sido reportado que los fosforamiditos que se activan con 1H-tetrazol reaccionan con la posición O6 de la guanosina.[73] Después de la oxidación con I2/ agua, el producto secundario, posiblemente a través de la migración de O6-N7, pasa a la depurinación. Los sitios apurinicos que se forman, se rompen fácilmente al final de la desprotección del oligonucleótido bajo las condiciones básicas (ver abajo) para dar lugar a oligonucleótidos más cortos y de esta forma reducir el rendimiento del producto completado. Las modificaciones de O6 son removidas rápidamente por un tratamiento con el reactivo de taponado siempre y cuando el paso del taponado se lleve a cabo antes de la oxidación con I2/ agua.
- La síntesis de fosfotrioatos de oligonucleótidos (OPS, ver abajo) no se involucra con la oxidación con I2/agua, y por lo tanto no sufre los efectos secundarios de la reacción que se describe anteriormente. Por otro lado, si el paso de taponado se lleva a cabo antes de una sulfurización, el soporte sólido puede contener los residuos del anhídrido acético y de N-metilimidazol que quedaron después del paso de taponado. La mezcla de taponado, interfiere con la reacción de transferencia de sulfuro, que resulta en la formación de los enlaces internucleosídicos de fosfato triester en el lugar de los triesteres PS deseados. Por lo tanto, para la síntesis de OPS, es recomendado llevar a cabo el paso de sulfurización antes del paso de taponado.[74]
Paso 4: oxidación
El enlace fosfito tricoordinado que se acaba de formar no es natural y tiene estabilidad limitada bajo las condiciones de la síntesis de oligonucleótidos. El tratamiento del soporte unido al material con yodo y agua en presencia de una base débil (piridina, lutidina, o colidina) oxida el fosfito triester en un fosfato triester tetracoordinado, que es un precursor protegido de un enlace internucleosídico de fosfato diester que ocurre de manera natural. La oxidación puede llevarse a cabo bajo condiciones anhídridas usando terc-butil hidroperoxido[75] o, más eficiente, (1S)-(+)-(10-camphorsulfonyl)-oxaziridino (CSO).[76][77][78] El paso de oxidación es sustituido con un paso de sulfurización para obtener fosfotrioatos de oligonucleótido (ver fosfotrioatos de oligonucleótido y su síntesis en la parte de abajo). En el último caso, el paso de sulfurización es mejor si se da antes del taponado.
Soporte sólido
En la síntesis de la fase sólida, el oligonucleótido es ensamblado mediante enlaces covalentes, a través de su terminal 3' del grupo hidroxilo, a un soporte soólido de material y restos unido a él durante todo el curso de la cadena de ensamblado. El soporte sólido está contenido en columnas cuyas dimensiones dependen en la escala de la síntesis y pueden variar entre 0.05mL y varios litros. La mayoría de los oligonucleótidos son sintetizados en una escala pequeña que va de los 40nmol a 1μmol. Recientemente, la síntesis compleja de los oligonucleótidos en donde el soporte sólido se encuentra en los pozos de las placas de múltiples pozos (comúnmente, 96 o 384 pozos por placa) se ha convertido en una opción para la síntesis paralela de oligonucleótidos en pequeña escala.[79] Al final de la cadena de ensamblado, los oligonucleótidos son liberados del soporte sólido y son eluidos de la columna o del pozo.
Material de soporte sólido
En contraste con la síntesis de la fase sólida orgánica y la síntesis peptídica, la síntesis de oligonucleótidos procede mejor en soportes sólidos no hinchables. Los dos materiales más usados en la fase sólida son el vidrio de poro controlado (CPG) y el poliestireno macroporoso (MPPS).[80]
- CPG es definido comúnmente por su tamaño de poro. En la química de oligonucleótidos, los tamaños de poros de 500, 1000, 1500, 2000 y 3000 Å se usan para permitir la preparación de 50, 80, 100, 150 y 200 oligonucleótidos, respectivamente. Para hacer que el CPG sea adecuado para el procesamiento, la superficie del material es tratada con (3-aminopropil) trietoxisilano para dar CPG al aminopropil. El brazo del aminopropil puede ser extendido para dar lugar a una cadena larga conocida como aminoalkil (LCAA) CPG. El grupo amino se usa como un punto adecuado de anclaje para los enlazadores en la síntesis de oligonucleoótidos (ver abajo)
- MPPS es adecuado para la síntesis de oligonucleótidos cuando son poco hinchables, el poliestireno altamente reticulado es obtenido por la polimerización de divinilbenceno (min 60%), estireno y 4-clorometilestireno en la presencia de un agente poroso. Los macroporos de clorometil MPPS obtenidos se convierten en aminometil MPPS.
Química enlazadora
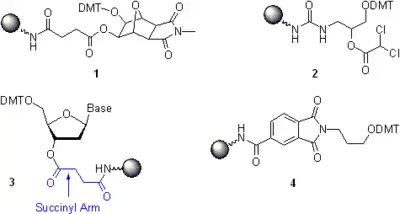
Para que el material del soporte sólido sea adecuado para la síntesis de oligonucleótidos, los enlazadores no nucleosidicos o los succionatos de nucleosido deben de estar unidos por un enlace covalente a un grupo amino reactivo, como aminopropil CPG, LCAA CPG o aminometil MPPS. El resto de los grupos aminos que no reaccionaron son cubiertos con anhídrido acético. Hay tres diferentes grupos de soportes de sólidos que se usan normalmente.
- Soportes universales. En un método más reciente, más conveniente y más usado, la síntesis empieza con el soporte universal, en donde un enlazador no nucleosídico es ligado al material del soporte del sólido (compuestos 1 y 2). Un fosforamito respectivo al residuo de nucleósido de 3'-terminal es acoplado al soporte sólido universal en el primer ciclo de síntesis en el ensamblaje del oligonucleótido usando los protocolos estándar. Después de esto el ensamblaje de cadena continua hasta que es completado y entonces el sólido de soporte es unido al oligonucleótido es desprotegido. La característica distintiva del sólido de soporte universal es que la liberación de los oligonucleótidos ocurre debido a la escisión hidrolítica de un enlace P-O que conecta el 3'-O de la 3'-terminal del residuo del nucleótido al enlazador universal como se muestra en el esquema 6. La ventaja crítica de este enfoque es que el mismo sólido de soporte es usado sin importar la secuencia de los oligonucleótidos que se desean sintetizar. Para remover por completo el enlazador y el fosfato 3'-terminal del oligonucleótido ensamblado, el sólido de soporte 1 y varios sólidos de soporte similares [81] requieren amonio gaseoso,[82] hidróxido de amonio acuoso, metilamina acuosa,[83] o su mezcla[84] y están comercialmente disponibles .[85][86] El soporte sólido 2[87] requiere una solución de amonia en metanol anhídrido y también está comercialmente disponible.[88][89]
- Soportes sólidos nucleosídicos. En un primer acercamiento, el grupo 3'hidroxilo del residuo del nucleosido 3'-terminal se une al soporte sólido a través de 3'-O-succinil como un compuesto 3. El ensamblaje de la cadena del oligonucleótido empieza con la unión de un bloque de construcción de fosforamidita al segundo residuo de nucleótido de 3'-terminal. El grupo hidroxilo 3'-terminal en oligonucleótidos sintetizados en soportes de sólido nucleosídicos son desprotegidos bajo ciertas condiciones, que son más ligeras que aquellas que aplican para los soportes de sólidos universales. Sin embargo, el hecho de que un soporte de sólido nucleosídico tenga que ser seleccionado en una secuencia específica reduce la complejidad del proceso sintético e incrementa la posibilidad del error humano.
- Soportes sólidos especiales. Son usados como para la adjunción de los grupos funcionales deseados en la 3'-terminal de los oligonucleótidos sintéticos. Por ejemplo, el soporte de sólido comercial[90] el soporte sólido 4[91] permite la preparación de oligonucleótidos que contienen en su 3'-terminal, enlazador 3-aminopropil. De manera similar a los fosforamiditos no nucleosídicos, hay otros soportes de sólidos diseñados para la fijación de grupos funcionales reactivos, grupos locutores no reactivos y modificadores terminales (por ejemplo, colesterol o otras ataduras hidrofóbicas) y adecuados a varias aplicaciones que están comercialmente disponibles. Información más detallada en varios soportes de sólidos para la síntesis de oligonucleótidos pueden encontrarse en un repaso reciente.[79]

Fosforotioatos de oligonucleótidos y su síntesis
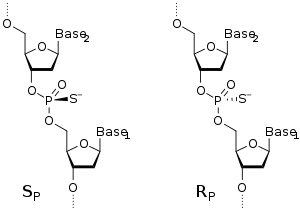
Los fosforotioatos de oligonucleótidos (OPS) son oligonucleótidos modificados en donde uno de los átomos de oxígeno en los restos del fosfato es reemplazado por un azufre. Solamente los fosforotioatos que tienen sulfuro en una posición en la que no se juntan, como se muestra en la figura, son muy usados y están disponibles comercialmente. El reemplazo del oxígeno que no une con el sulfuro, crea un nuevo centro quiral en el fósforo. En el caso de un dinucleótido, esto resulta en la formación de un diasterómero de monofosforotioatos Sp y Rp dinucleósidos, cuyas estructuras se muestran en la figura. En un oligonucleótido n-mer donde todos los (n-1) enlaces internucleosídicos son enlaces de fosforotioato, el número de diasterómeros m es calculado com m = 2(n – 1). Como son análogos de ácidos nucleicos no naturales, los OPS son sustancialmente más estables en la hidrólisis de nucleasas, las enzimas que se encargan de destruir ácidos nucleicos al romper los puentes del enlace P-O en la mitad del fosfodiester. Esta propiedad determina el uso de OPS como oligonucleótidos antisentido in vitro y en vivo, aplicaciones donde la amplia exposición a las nucleasas es inevitable. De manera similar, para mejorar la estabilidad de los siRNAs, al menos un enlace de fosforotiato es introducido en la terminal 3' en las hembras de ambos sentidos y de antisentido. En cuanto a la quiralidad, los OPS puros, todos los diasterómeros Sp son más estables a la degradación enzimática que sus análogos Rp.[92] Sin embargo la preparación de la quiralidad de los OPS puros sigue siendo un reto sintético.[12][93] En las prácticas de laboratorio, diferentes mezclas de diasterómeros de OPS son usadas normalmente.
La síntesis de los OPS es muy parecida a la de los oligonucleótidos naturales. La diferencia es que el paso de oxidación es reemplazado por una transferencia de sulfuro (sulfuración) y que el sistema de taponado es llevado a cabo hasta después de la sulfuración. Solo hay 3 reactivos reportados capaces de llevar a cabo la transferencia de sulfuro eficiente y que estén comercialmente disponibles:
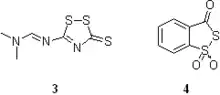
- 3-(Dimethylaminomethylidene)amino)-3H-1,2,4-dithiazole-3-thione, DDTT (3) provee cinética rápida de sulfuración y alta estabilidad en la solución.[74][94][95] El reactivo está disponible de varias fuentes.[96][97]
- 3H-1,2-benzodithiol-3-one 1,1-dioxide (4)[98][99] también conocido con el reactivo de Beaucage, tiene una mejor solubilidad en acetonitrilo y tiempos cortos de reacción. Sin embargo el reactivo en solución tiene una estabilidad limitada y es menos eficiente en los enlaces de sulforización de RNA.[94][95]
- N,N,N'N'-Tetraethylthiuram disulfide (TETD) es soluble en acetonitrilo y es comercialmente disponible.[100] Sin embargo, la reacción de sulfurización de un enlace internucleosídico de DNA con TETD requiere 15 minutos,[101] lo que es más de 10 veces más lento que en los compuestos 3 y 4.
Automatización
En el pasado, la síntesis de oligonucleótidos se llevaba a cabo manualmente en solución o en fase sólida. La síntesis de la fase sólida fue implementada usando, como contenedores de la fase sólida, columnas de vidrio miniatura similares en la forma de columnas de cromatografía de baja presión o jeringas con filtros porosos.[102] Actualmente la síntesis de oligonucleótidos en fase sólida se lleva a cabo de manera automática usando instrumentos controlados por computadora (sintetizadores de oligonucleótidos) y son técnicamente implementados en formatos de columna, placa de pozos múltiples, y de arreglos. El formato de columna funciona mejor en la investigación y en aplicaciones a grande escala, cuando la síntesis no es compleja.[103] El formato de la placa de pozos múltiples está diseñado específicamente para la síntesis compleja en una escala pequeña para satisfacer la creciente demanda de la industria y académica para la síntesis de oligonucleótidos.[104] Se encuentran disponibles comercialmente un número de sintetizadores de oligonucleótidos para una síntesis a pequeña escala[105][106][107][108][109] y también para la síntesis de mediana a larga escala[110]
Síntesis de medio a largo plazo de la síntesis de oligonucleótidos - Historia
Los sintetizadores de oligonucleótidos a larga escala eran desarrollados con frecuencia aumentando las capacidades de una plataforma de instrumentos preexistente. En 1980 surgió uno de los primeros sintetizadores a mediana escala, fue fabricado por la compañía Biosearch, en Novato, CA (8800). Esta plataforma fue originalmente diseñada como un sintetizador de péptidos y su usaba un reactor de lecho de fluido esencial para el alojamiento de las características de hinchamiento de poliestireno en los soportes utilizados en la metodología de Merrifield. La síntesis de oligonucleótidos involucra el uso de CPG (vidrio de poro controlado) que es un soporte rígido más adecuado para los reactores de columna que se describen en la parte de arriba. La escala de 8800 estaba limitada al flujo requerido para fluidizar el soporte. Algunos de los diseños de los reactores de novel, así como que las presiones estuvieran por arriba de lo normal permitían al 8800 alcanzar escalas que podían preparar 1mmol de oligonucleótidos. A mediados de los años 1990, varias compañías desarrollaron plataformas basadas en líquidos de cromatografía preparativas y semi-preparativas. Estos sistemas eran adecuados para un enfoque de reactor de columna. En la mayoría de los casos lo único que se requería era aumentar el número de fluidos que podían ser entregados a una columna. La oligo síntesis requiere un mínimo de 10 y los líquidos de cromatografía normalmente acomodan 4. Esto era una tarea de diseño fácil y algunas estrategias semi automáticas funcionaban sin hacer ninguna modificación al equipo LC que ya existía. PerSeptive Biosystems y Pharmacia (GE) eran 2 de las muchas compañías que desarrollaron sintetizadores a partir de cromatografías líquidas. Genomic Technologies, Inc.[111] era una de las pocas compañías que desarrollo sintetizadores de oligonucleótidos a larga escala. La plataforma inicial llamada VLSS que significa sintetizadores a muy grande escala por sus siglas en inglés, utilizaba columnas de cromatografía líquida como reactores y podía sintetizar hasta 75 milimoles de material. Muchas fábricas de síntesis de oligonucleótidos diseñaban y producían sus propias plataformas y por eso se conoce poco de estas. El diseño VLSS continuó perfeccionándose y continúa en el sintetizador Qmaster [112] que es una plataforma a pequeña escala que produce cantidades de miligramos a gramos de oligonucleótidos sintetizados. Las prácticas actuales de la síntesis de los oligonucleótidos químicamente modificados a larga escala han sido revisados recientemente.[113]
Síntesis de los microarreglos de oligonucleótidos
El microarreglo de un oligonucleótido puede ser visto como una placa de pozos múltiples, en donde las paredes que dividen los pozos son removidos intencionalmente. Con respecto a la parte química, la síntesis de los microarreglos de oligonucleótidos es diferente a la síntesis de oligonueclótidos convencional en dos cosas principales:
- Los oligonucleótidos permanecen unidos a la fase sólida de manera permanente, lo que requiere el uso de enlazadores que sean estables bajo las condiciones del final del procedimiento de desprotección.
- Es necesario usar técnicas de sitio selectivo de 5'-desprotección al considerar las siguientes razones: la ausencia de divisores físicos entre los sitios ocupados por los oligonucleótidos individuales, un espacio muy limitado en la superficie del microarreglo (una secuencia de oligonucleótido ocupa un cuadrado de 25×25 μm)[114] y el requerimiento de alta fidelidad de la síntesis del oligonucleótido. En un enfoque, cuando el grupo 5'-O-ODMT es removido, se ve efectuado por una generación electroquímica del ácido en los sitios requeridos.[115] Otro enfoque, usa 5'-O-(a-metil-6-oxicarbonilo piperonilo nitro) (MeNPOC) como grupo protector, el cual puede ser removido con radiación de luz UV a 365 nm de longitud de onda.[114]

Procesamiento post-sintético
Después de que el ensamblaje de la cadena es completado, el soporte sólido unido al oligonucleótido está totalmente protegido:
- El grupo 5'-terminal 5'-hidroxilo es protegido por el grupo DMT;
- Los restos del fosfato internucleosídico o fosforotrioato están protegidos por los grupos 2-cianoétilo.
- Los grupos amino exocíclicos en todas las bases nitrogenadas, excepto en T y U, están protegidos por grupos protectores acilo.
Para que el oligonucleótido sea funcional, todos los grupos protectores deben ser removidos. La base N-acil de protección y el 2-cianoetil fosfato de protección pueden ser removidos simultáneamente con el tratamiento de bases inorgánicas o de aminas. Sin embargo, la aplicabilidad de este método está limitada por el hecho de que la hendidura del 2-cianoetilo fosfato de protección lleva a la formación de acrilonitrilo como un producto secundario. Bajo las condiciones básicas que se requieren para remover la protección del N-acilo, el acrilonitrilo es capaz de una alquilación de bases nitrogenadas, principalmente, en la posición N3 de los residuos de la timina y el uracilo para dar lugar a los respectivos aductos de N3-(2-cianoetil) a través de la reacción de Michael. La formación de estos productos secundarios puede ser evitada tratando a los oligonucleótidos unidos al soporte sólido con soluciones de bases en un solvente orgánico, por ejemplo, con 50% trietilamina en acetonitrilo[116] o con 10% de dietilamina en acetonitrilo.[117] Este tratamiento es muy recomendado para las preparaciones de mediana y gran escala y es opcional para la síntesis de pequeña escala, donde la concentración generada de acrilonitrilo en la desprotección es baja.
Independientemente de si los grupos protectores de fosfato son removidos primero, los oligonucleótidos unidos al soporte sólido son desprotegidos usando uno de los dos enfoques generales:
- (1) A menudo, el grupo 5'-DMT es removido al final del ensamblaje de la cadena de oligonucleoótidos. Los oligonucleótidos son liberados de la fase sólida y desprotegidos (base y fosfato) por tratamiento con hidróxido de amonio acuoso, metilamina acuosa, sus mezclas,[41] amonio gaseoso o metilaminane[118] o, menos común, soluciones de otras aminas primarias o álcalis a temperatura ambiente o elevada. Esto remueve toda la protección que quedaba en los grupos de 2'-desoxioligonucleótidos, resultando una mezcla que contiene el producto deseado. Si los oligonucleótidos contienen residuos de ribonuleótidos 2'-O-protegidos, el protocolo para la desprotección incluye un segundo paso en el que el 2'-O-protector de grupos sillyl son removidos mediante un tratamiento con ion floruro a través de varios métodos.[119] El producto totalmente desprotegido puede ser usado así, o el oligonucleótido puede ser purificado por varios métodos. El más común es cuando el producto crudo es desalinizado usando precipitación de etanol, cromatografía por exclusión de tamaño o HPLC de fase reversa. Para eliminar productos de truncación no requeridos, los oligonucleótidos pueden ser purificados mediante electroforesis en gel de poliacrilamida o de intercambio aniónico HPLC seguido de desalinización.
- (2) El segundo enfoque solamente es usado cuando el método de purificación es HPLC de fase reversa. En este caso, el grupo 5'-terminal DMT que sirve como manija hidrofóbica para purificación, permanece hasta el final de la síntesis. El oligonucleótido es desprotegido bajo las condiciones básicas descritas anteriormente y a partir de una evaporación, es purificado mediante una HPLC de fase reversa. El material recolectado es posteriormente detritilación bajo condiciones ácidas acuosas. En una pequeña escala (menos de 0.01-0.02mmol), se usa el tratamiento con 80% de ácido acético acuoso durante 15-30 minutos a temperatura ambiente, seguido por una evaporación de la mezcla. Finalmente el producto es desalinizado, como fue descrito anteriormente.
- Para algunas aplicaciones, grupos informadores adicionales pueden estar unidos a un oligonucleótido usando una variedad de procedimientos post-sintéticos.
Caracterización

Así como con cualquier otro compuesto orgánico, es prudente caracterizar oligonucleótidos sintéticos según su preparación. En casos más complejos (investigación y síntesis a gran escala) los oligonucleótidos se caracterizan por su desprotección y su purificación. A pesar de que el enfoque principal de la caracterización es la secuenciación, una rutina relativamente económica, las consideraciones en la reducción de costos evitan su uso en la rutina de la fabricación de oligonucleótidos. En la práctica del día a día, es suficiente con obtener la masa molecular de un oligonucleótido mediante el registro de su espectro de masa. Hay dos métodos que son usados en la caracterización de oligonucleótidos: electrospray de espectometría de masa (ES MS) y láser asistido por matriz de desorción / espectrometría de masas de ionización de tiempo de vuelo (MALDI-TOF). Para obtener el informe de espectro, es muy importante cambiar todos los iones de metal que puedan estar presentes en la muestra de iones de amonio o de tetraalquilamonio antes de someter una muestra al análisis por cualquiera de los métodos mencionados.
- En un espectro ES MS, un oligonucleótido en específico genera una serie de iones que corresponden a diferentes estados de ionización del compuesto. En consecuencia, el oligonucleoótido con masa molecular M genera iones con masas (M-nH)/n donde M es la masa molecular del oligonucleótido en forma de un ácido libre (todas las cargas negativas de los grupos fosfodiéster internucleosídicos son neutralizados con H+), n es el estado de ionización y H es la masa atómica del hidrógeno (1 Da). Los iones con un rango n de 2 a 5 son más útiles para la caracterización. Recientemente el software suministrado con los instrumentos fabricados es capaz de llevar a cabo un procedimiento de deconvolución, lo que significa que encuentra picos de iones que pertenecen al mismo conjunto y que deriva la masa molecular del oligonucleótido.
- Para obtener información más detallada de la impureza del perfil de los oligonucleótidos se usa la cromatografía líquida de masa (LC-MS o HPLC-MS)[120] o electroforesis capilar de espectometría de masa(CEMS)[121]
Referencias
- Beaucage, S.; Iyer, R. (1992). «Advances in the Synthesis of Oligonucleotides by the Phosphoramidite Approach». Tetrahedron 48: 2223. doi:10.1016/S0040-4020(01)88752-4.
- Brown, D. M. A brief history of oligonucleotide synthesis. Methods in Molecular Biology (Totowa, NJ, United States) (1993), 20 (Protocols for Oligonucleotides and Analogs), 1–17.
- Reese, Colin B. (2005). «Oligo- and poly-nucleotides: 50 years of chemical synthesis». Organic & Biomolecular Chemistry 3 (21): 3851. doi:10.1039/b510458k.
- Iyer, R. P.; Beaucage, S. L. 7.05. Oligonucleotide synthesis. In: Comprehensive Natural Products Chemistry, Vol. 7: DNA and Aspects of Molecular Biology. Kool, Eric T.; Editor. Neth. (1999), Elsevier, Amsterdam, pp. 105–152.
- Michelson, A. M.; Todd, A. R. (1955). «Nucleotides part XXXII. Synthesis of a dithymidine dinucleotide containing a 3′: 5′-internucleotidic linkage». J. Chem. Soc.: 2632. doi:10.1039/JR9550002632.
- Hall, R. H.; Todd, A.; Webb, R. F. (1957). «644. Nucleotides. Part XLI. Mixed anhydrides as intermediates in the synthesis of dinucleoside phosphates». J. Chem. Soc.: 3291. doi:10.1039/JR9570003291.
- Froehler, B. C.; Ng, P. G.; Matteucci, M. D. (1986). «Synthesis of DNA via deoxynucleoside H-phosphonate intermediates». Nucl. Acids Res. 14 (13): 5399-5407. PMC 311548. PMID 3737406. doi:10.1093/nar/14.13.5399.
- Garegg, P. J.; Lindh, I.; Regberg, T.; Stawinski, J.; Strömberg, R. (1986). «Nucleoside H-phosphonates. III. Chemical synthesis of oligodeoxyribonucleotides by the hydrogenphosphonate approach». Tetrahedron Lett. 27 (34): 4051. doi:10.1016/S0040-4039(00)84908-4.
- Agrawal, S.; Goodchild, J.; Civeira, M. P.; Thornton, A. H.; Sarin, P. S.; Zamecnik, P. C. (1988). «Oligodeoxynucleotide phosphoramidates and phosphorothioates as inhibitors of human immunodeficiency virus». Proc. Natl. Acad. Sci. U.S.A. 85 (19): 7079-7083. Bibcode:1988PNAS...85.7079A. doi:10.1073/pnas.85.19.7079.
- Kamer, P. C. J.; Roelen, H. C. P. F.; Van den Elst, H.; Van der Marel, G. A.; Van Boom, J. H. (1989). «An efficient approach toward the synthesis of phosphorothioate diesters via the Schoenberg reaction». Tetrahedron Lett. 30 (48): 6757-6760. doi:10.1016/S0040-4039(00)70669-1.
- Agrawal, S.; Tang, J. Y. (1990). «Efficient synthesis of oligoribonucleotide and its phosphorothioate analog using H-phosphonate approach». Tetrahedron Lett. 31 (52): 7541-7544. doi:10.1016/S0040-4039(00)97293-9.
- Almer, H.; Stawinski, J.; Strӧmberg, R. (1996). «Solid support synthesis of all-Rp-oligo(ribonucleoside phosphorothioate)s.». Nucleic Acids Res. 24 (19): 3811-3820. doi:10.1093/nar/24.19.3811.
- Tram, K.; Wang, X.; Yan, H. (2007). «Facile Synthesis of Oligonucleotide Phosphoroselenoates». Org. Lett. 9 (24): 5103-5106. doi:10.1021/ol702305v.
- Froehler, B. C. (1986). «Deoxynucleoside H-phosphonate diester intermediates in the synthesis of internucleotide phosphate analogs». Tetrahedron Lett. 27 (46): 5575-5578. doi:10.1016/S0040-4039(00)85269-7.
- Froehler, B. C.; Ng, P. G.; Matteucci, M. D. (1988). «Phosphoramidate analogs of DNA: synthesis and thermal stability of heteroduplexes». Nucleic Acids Res. 16 (11): 4831-4839. doi:10.1093/nar/16.11.4831.
- Dagle, J. M.; Andracki, M. E.; DeVine, R. J.; Walder, J. (1991). «Physical properties of oligonucleotides containing phosphoramidate-modified internucleoside linkages». Nucleic Acids Res. 19 (8): 1805-1810. doi:10.1093/nar/19.8.1805.
- Maier, M. A.; Guzaev, A. P.; Manoharan, M. (2000). «Synthesis of Chimeric Oligonucleotides Containing Phosphodiester, Phosphorothioate, and Phosphoramidate Linkages». Org. Lett. 2 (13): 1819-1822. doi:10.1021/ol005842h.
- Mohe, N. U.; Padiya, K. J.; Salunkhe, M. M. (2003). «An efficient oxidizing reagent for the synthesis of mixed backbone oligonucleotides via the H-Phosphonate approach». Bioorg. Med. Chem. 11 (7): 1419-1431. doi:10.1016/S0968-0896(02)00615-6.
- Wada, T.; Sato, Y.; Honda, F.; Kawahara, S.; Sekine, M. (1997). «Chemical Synthesis of Oligodeoxyribonucleotides Using N-Unprotected H-Phosphonate Monomers and Carbonium and Phosphonium Condensing Reagents: O-Selective Phosphonylation and Condensation». J. Amer. Chem. Soc. 119 (52): 12710-12721. doi:10.1021/JA9726015.
- Kung, P. P. and Jones, R. A. (1992). «H-phosphonate DNA synthesis without amino protection». Tetrahedron Lett. 33 (40): 5869-5872. doi:10.1016/S0040-4039(00)61075-4.
- Gilham, P. T.; Khorana, H. G. (1958). «Studies on Polynucleotides. I. A New and General Method for the Chemical Synthesis of the C5″-C3″ Internucleotidic Linkage. Syntheses of Deoxyribo-dinucleotides». Journal of the American Chemical Society 80: 6212. doi:10.1021/ja01556a016.
- Letsinger, R. L.; Ogilvie, Kelvin K. (1969). «Nucleotide chemistry. XIII. Synthesis of oligothymidylates via phosphotriester intermediates». Journal of the American Chemical Society 91: 3350. doi:10.1021/ja01040a042.
- Reese, C. (1978). «The chemical synthesis of oligo- and poly-nucleotides by the phosphotriester approach». Tetrahedron 34: 3143. doi:10.1016/0040-4020(78)87013-6.
- Efimov, V. A.; Buryakova, A. A.; Reverdatto, S. V.; Chakhmakhcheva, O. G.; Ovchinnikov, Yu. A. (1983). «Rapid synthesis of long-chain deoxyribooligonucleotides by the N-methylimidazolide phosphotriester method». Nucleic Acids Res 11 (23): 8369-8387. PMC 326588. PMID 6324083. doi:10.1093/nar/11.23.8369.
- Efimov, V. A; Molchanova, N. S.; Chakhmakhcheva, O. G. (2007). «Approach to the synthesis of natural and modified oligonucleotides by the phosphotriester method using O-nucleophilic intramolecular catalysis.». Nucleosides, nucleotides & nucleic acids 26 (8-9): 1087-93. ISSN 1525-7770. PMID 18058542. doi:10.1080/15257770701516268.
- Letsinger, R. L.; Mahadevan, V (1966). «Stepwise synthesis of oligodeoxyribonucleotides on an insoluble polymer support.». Journal of the American Chemical Society 88 (22): 5319. PMID 5979268. doi:10.1021/ja00974a053.
- Letsinger, R. L.; Finnan, J. L.; Lunsford, N. B. (1975). «Nucleotide chemistry. XX. Phosphite coupling procedure for generating internucleotide links». J. Amer. Chem. Soc. 97 (11): 3278-9. PMID 1133350. doi:10.1021/ja00844a090.
- McBride, L. J.; Caruthers, M. H. (1983). «Nucleotide chemistry. X. An investigation of several deoxynucleoside phosphoramidites useful for synthesizing deoxyoligonucleotides». Tetrahedron Lett. 24 (3): 245-248. doi:10.1016/S0040-4039(00)81376-3.
- Adams, S. P.; Kavka, K. S.; Wykes, E. J.; Holder, S. B.; Galluppi, G. R. (1983). «Hindered dialkylamino nucleoside phosphite reagents in the synthesis of two DNA 51-mers». J. Amer. Chem. Soc. 105 (3): 661-663. doi:10.1021/ja00341a078.
- Matteucci, M. D.; Caruthers, M. H. (1981). «Synthesis of deoxyoligonucleotides on a polymer support». J. Amer. Chem. Soc. 103 (11): 3185. doi:10.1021/ja00401a041.
- Beaucage, S. L.; Caruthers M. H. (1981). «Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis». Tetrahedron Lett. 22 (20): 1859. doi:10.1016/S0040-4039(01)90461-7.
- Sinha, N. D.; Biernat, J.; McManus, J.; Kӧster, H. (1984). «Polymer support oligonucleotide synthesis. XVIII: use of β-cyanoethyl-N,N-dialkylamino-/N-morpholino phosphoramidite of deoxynucleosides for the synthesis of DNA fragments simplifying deprotection and isolation of the final product». Nucl. Acids Res. 12 (11): 4539-4557. PMC 318857. PMID 6547529. doi:10.1093/nar/12.11.4539.
- Beaucage, S. L.; Iyer, R. P. (1992). «Advances in the Synthesis of Oligonucleotides by the Phosphoramidite Approach». Tetrahedron 48 (12): 2223. doi:10.1016/S0040-4020(01)88752-4.
- «Beta-Cyanoethyl Phosphoramidites». Products.appliedbiosystems.com. Consultado el 12 de mayo de 2009.
- «Biosearch Technologies». Biosearchtech.com. Consultado el 12 de mayo de 2009.
- «ChemGenes Corporation, a Biotechnology company». Chemgenes.com. Consultado el 12 de mayo de 2009.
- Powell, M. (17 de enero de 2008). «Applied Biosystems Instruments». Glenresearch.com. Consultado el 12 de mayo de 2009.
- Sekine, M. DNA synthesis without base protection. In: Current protocols in nucleic acid chemistry. Beaucage, S. L., Editor (John Wiley & Sons, Inc.) (2004), Chapter 3, Unit 3.10.,pp. 3.10.1-3.10.15. PubMed ID:18428925
- Gryaznov, S. M.; Letsinger, R. L. (1991). «Synthesis of oligonucleotides via monomers with unprotected bases». J. Amer. Chem. Soc. 113 (15): 5876-5877. doi:10.1021/ja00015a059.
- Sekine, M., Ohkubo, A., and Seio, K. (2003). «Protonblock strategy for the synthesis of oligodeoxynucleotides without base protection, capping reaction, and P-N bond cleavage reaction». J. Org. Chem. 68 (14): 5478-5492. doi:10.1021/jo034204k.
- Reddy, M. P.; Hanna, N. B.; Farooqui, F (1997). «Ultrafast Cleavage and Deprotection of Oligonucleotides Synthesis and Use of CAc Derivatives». Nucleosides & Nucleotides 16 (7): 1589-1598. doi:10.1080/07328319708006236.
- McMinn, D. L.; Greenberg, M. M. (1997). «Synthesis of oligonucleotides containing 3'-alkyl amines using N-isobutyryl protected deoxyadenosine phosphoramidite». Tetrahedron Lett. 38 (18): 3123. doi:10.1016/S0040-4039(97)00568-6.
- Schulhof, J. C.; Molko, D.; Teoule, R. (1987). «The final deprotection step in oligonucleotide synthesis is reduced to a mild and rapid ammonia treatment by using labile base-protecting groups». Nucleic Acids Res. 15 (2): 397-416. PMC 340442. PMID 3822812. doi:10.1093/nar/15.2.397.
- Zhu, Q.; Delaney, M. O.; Greenberg, M. M. (2001). «Observation and elimination of N-acetylation of oligonucleotides prepared using fast-deprotecting phosphoramidites and ultra-mild deprotection». Bioorg. & Med. Chem. Lett. 11 (9): 1105. doi:10.1016/S0960-894X(01)00161-5.
- McBride, L. J.; Kierzek, R.; Beaucage, S. L.; Caruthers, M. H. (1986). «Nucleotide chemistry. 16. Amidine protecting groups for oligonucleotide synthesis». J. Amer. Chem. Soc. 108 (8): 2040-2048. doi:10.1021/ja00268a052.
- Guzaev, A. P.; Manoharan, M. (2001). «Phosphoramidite Coupling to Oligonucleotides Bearing Unprotected Internucleosidic Phosphate Moieties». J. Org. Chem. 66 (5): 1798-1804. PMID 11262130. doi:10.1021/jo001591e.
- Ogilvie, K. K.; Theriault, N.; Sadana, K. L. (1977). «Synthesis of oligoribonucleotides». J. Amer. Chem. Soc. 99 (23): 7741-7743. doi:10.1021/ja00465a073.
- Usman, N.; Ogilvie, K. K.; Jiang, M. Y.; Cedergren, R. J. (1987). «The automated chemical synthesis of long oligoribuncleotides using 2'-O-silylated ribonucleoside 3'-O-phosphoramidites on a controlled-pore glass support: synthesis of a 43-nucleotide sequence similar to the 3'-half molecule of an Escherichia coli formylmethionine tRNA». J. Amer. Chem. Soc. 109 (25): 7845-7854. doi:10.1021/ja00259a037.
- Usman, N.; Pon, R. T.; Ogilvie, K. K. (1985). «Preparation of ribonucleoside 3'-O-phosphoramidites and their application to the automated solid phase synthesis of oligonucleotides». Tetrahedron Lett. 26 (38): 4567-4570. doi:10.1016/S0040-4039(00)98753-7.
- Scaringe, S. A.; Francklyn, C.; Usman, N. (1990). «Chemical synthesis of biologically active oligoribonucleotides using β-cyanoethyl protected ribonucleoside phosphoramidites». Nucl. Acids Res. 18 (18): 5433-5441. doi:10.1093/nar/18.18.5433.
- Pitsch, S.; Weiss, P. A.; Wu, X.; Ackermann, D.; Honegger, T. (1999). «Fast and reliable automated synthesis of RNA and partially 2'-O-protected precursors ("caged RNA") based on two novel, orthogonal 2'-O-protecting groups». Helv. Chim. Acta 82 (10): 1753-1761. doi:10.1002/(SICI)1522-2675(19991006)82:10<1753::AID-HLCA1753>3.0.CO;2-Y.
- Pitsch, S.; Weiss, P. A.; Jenny, L.; Stutz, A.; Wu, X. (2001). «Reliable chemical synthesis of oligoribonucleotides (RNA) with 2'-O-[(triisopropylsilyl)oxy]methyl(2'-O-tom)-protected phosphoramidites». Helv. Chim. Acta 84 (12): 3773-3795. doi:10.1002/1522-2675(20011219)84:12<3773::AID-HLCA3773>3.0.CO;2-E.
- Guzaev, A.; Salo, H.; Azhayev, A.; Lӧnnberg, H. (1995). «A new approach for chemical phosphorylation of oligodeoxyribonucleotides at the 5'-terminus». Tetrahedron 51 (34): 9375-9384. doi:10.1016/0040-4020(95)00544-I.
- Sinha, N. D.; Cook, R. M. (1988). «The preparation and application of functionalized synthetic oligonucleotides: III. Use of H-phosphonate derivatives of protected amino-hexanol and mercapto-propanol or-hexanol». Nucl. Acids Res. 16 (6): 2659-2669. PMC 336396. PMID 3362678. doi:10.1093/nar/16.6.2659.
- Jones, D. S.; Hachmann, J. P.; Conrad, M. J.; Coutts, S.; Livingston, D. A. Intermediates for providing functional groups on the 5' end of oligonucleotides, (1995) Patente USPTO n.º 5391785.
- Podyminogin, M. A.; Lukhtanov, E. A.; Reed, M. W. (2001). «Attachment of benzaldehyde-modified oligodeoxynucleotide probes to semicarbazide-coated glass». Nucl. Acids Res. 29 (24): 5090-5098. PMC 97543. PMID 11812841. doi:10.1093/nar/29.24.5090.
- Lebedev, A. V.; Combs, D.; Hogrefe, R. I. (2007). «Preactivated Carboxyl Linker for the Rapid Conjugation of Alkylamines to Oligonucleotides on Solid Support». Bioconjugate Chem. 18 (5): 1530-1536. doi:10.1021/bc0603891.
- Alvira, M.; Eritja, R. (2007). «Synthesis of oligonucleotides carrying 5'-5' linkages using copper-catalyzed cycloaddition reactions». Chemistry & Biodiversity 4 (12): 2798-2809. doi:10.1002/cbdv.200790229.
- Brush, C. K. "Fluorescein Labelled Phosphoramidites". (1996) Patente USPTO n.º 5583236.
- Pitner, J. B.; Linn, C. P. "Synthesis and use of labelled phosphoramidite compositions". (2000) Patente USPTO n.º 6114518.
- Levenson; C.; Chang; C.-A; Oakes; F. T. "Oligonucleotide functionalizing reagents". (1990) Patente USPTO n.º 4914210.
- Durand, M.; Chevrie, K.; Chassignol, M.; Thuong, N. T.; Maurizot, J. C. (1990). «Circular dichroism studies of an oligodeoxyribonucleotide containing a hairpin loop made of a hexaethylene glycol chain: conformation and stability». Nucl. Acids Res. 18 (21): 6353-6359. PMC 332506. PMID 2243780. doi:10.1093/nar/18.21.6353.
- Christiano, A.; McSwiggen, J. "RNA interference-mediated inhibition of retinoblastoma (RB1) gene expression using short interfering nucleic acid". PCT Int. Appl. (2006), WO 2006078798 A2.
- Pon, R. T. (1991). «A long chain biotin phosphoramidite reagent for the automated synthesis of 5'-biotinylated oligonucleotides». Tetrahedron Lett. 32 (14): 1715-1718. doi:10.1016/S0040-4039(00)74311-5.
- Sproat, B.; Colonna, F.; Mullah, B.; Tsou, D.; Andrus, A.; Hampel, A.; Vinayak, R. (1995). «An efficient method for the isolation and purification of oligoribonucleotides». Nucleosides & Nucleotides 14 (1&2): 255-273. doi:10.1080/15257779508014668.
- Stutz, A.; Hobartner, C.; Pitsch, S. (2000). «Novel fluoride-labile nucleobase-protecting groups for the synthesis of 3'(2')-O-amino-acylated RNA sequences». Helv. Chim. Acta 83 (9): 2477-2503. doi:10.1002/1522-2675(20000906).
- Welz, R.; Muller, S. (2002). «5-(Benzylmercapto)-1H-tetrazole as activator for 2'-O-TBDMS phosphoramidite building blocks in RNA synthesis». Tetrahedron Lett. 43 (5): 795-797. doi:10.1016/S0040-4039(01)02274-2.
- Vargeese, C.; Carter, J.; Yegge, J.; Krivjansky, S.; Settle, A.; Kropp, E.; Peterson, K.; Pieken, W. (1998). «Efficient activation of nucleoside phosphoramidites with 4,5-dicyanoimidazole during oligonucleotide synthesis». Nucl. Acids Res. 26 (4): 1046-1050. PMC 147346. PMID 9461466. doi:10.1093/nar/26.4.1046.
- Wei, Xia (2013). «Coupling activators for the oligonucleotide synthesis via phosphoramidite approach». Tetrahedron 69 (18): 3615-3637. doi:10.1016/j.tet.2013.03.001.
- Ogilvie, K. K.; Usman, N.; Nicoghosian, K.; Cedergren, R. J. (1988). «Total chemical synthesis of a 77-nucleotide-long RNA sequence having methionine-acceptance activity». Proc. Natl. Acad. Sci. USA 85 (16): 5764-5768. Bibcode:1988PNAS...85.5764O. PMC 281845. PMID 3413059. doi:10.1073/pnas.85.16.5764.
- Wu, T.; Ogilvie, K. K.; Perreault, J. Pierre; Cedergren, R. J. (1989). «Convenient procedure for the preparation of specific mixed DNA-RNA polymers». J. Amer. Chem. Soc. 111 (22): 8531-8533. doi:10.1021/ja00204a043.
- Pon, R. T. (1987). «Enhanced coupling efficiency using 4-dimethylaminopyridine (DMAP) and either tetrazole, 5-(o-nitrophenyl)tetrazole, or 5-(p-nitrophenyl)tetrazole in the solid phase synthesis of oligoribonucleotides by the phosphoramidite procedure». Tetrahedron Lett. 28 (32): 3643-3646. doi:10.1016/S0040-4039(00)96344-5.
- Pon, R. T.; Usman, N.; Damha, M. J.; Ogilvie, K. K. (1986). «Prevention of guanine modification and chain cleavage during the solid phase synthesis of oligonucleotides using phosphoramidite derivatives». Nucl. Acids Res. 14 (16): 6453-6470. doi:10.1093/nar/14.16.6453.
- Guzaev, A. P. (2011). «Reactivity of 3H-1,2,4-dithiazole-3-thiones and 3H-1,2-dithiole-3-thiones as sulfurizing agents for oligonucleotide synthesis». Tetrahedron Lett. 52 (3): 434-437. doi:10.1016/j.tetlet.2010.11.086.
- Alul, R. H.; Singman, C. N.; Zhang, G.; Letsinger, R. L. (1991). «Oxalyl-CPG: a labile support for synthesis of sensitive oligonucleotide derivatives». Nucleic Acids Res. 19 (7): 1527-1532. doi:10.1093/nar/19.7.1527.
- «New Product: 0.5M CSO for non-aqueous oxidation in DNA synthesis». Glenres.com. Consultado el 28 de enero de 2013.
- Manoharan, M.; Lu, Y.; Casper, M. D.; Just, G. (2000). «Allyl Group as a Protecting Group for Internucleotide Phosphate and Thiophosphate Linkages in Oligonucleotide Synthesis: Facile Oxidation and Deprotection Conditions». Org. Lett. 2 (3): 243-246. doi:10.1021/ol9910518.
- Prakash, T. P.; Johnston, J. F.; Graham, M. J.; Condon, T. P.; Manoharan, M. (2004). «2'-O-[2-[(N,N-dimethylamino)oxy]ethyl]-modified oligonucleotides inhibit expression of mRNA in vitro and in vivo». Nucleic Acids Res. 32 (2): 828-833. doi:10.1093/nar/gkh220.
- Guzaev, A. P. Solid-phase supports for oligonucleotide synthesis. In: Current protocols in nucleic acid chemistry. (John Wiley & Sons, Inc.) (2013), Chapter 3, Unit 3.1., pp. 3.1.1-3.1.60. doi 10.1002/0471142700.nc0301s53
- Pon, R. T. Solid-phase supports for oligonucleotide synthesis. Methods in Molecular Biology (Totowa, NJ, United States) (1993), 20 (Protocols for Oligonucleotides and Analogs), 465–496 doi 10.1385/0-89603-281-7:465.
- Guzaev, A. P.; Manoharan, M. (2003). «A conformationally preorganized universal solid support for efficient oligonucleotide synthesis». J. Amer. Chem. Soc. 125 (9): 2380-1. PMID 12603111. doi:10.1021/ja0284613.
- Jensen, M. A.; Anderson, K. M.; Davis, R. W. (2010). «Gas-Phase Cleavage and Dephosphorylation of Universal Linker-Bound Oligodeoxynucleotides». Nucleosides, Nucleotides and Nucl. Acids 29 (11): 867-878. doi:10.1080/15257770.2010.534757.
- «Glen Research Report of Products for RNA and DNA Oligonucelotide Synthesis, Modification and Labelling». Glenresearch.com. 17 de enero de 2008. Consultado el 12 de mayo de 2009.
- «AM Chemicals, LLC, a supplier of solid supports and reagents for oligonucleotide and organic synthesis on solid phase». Amchemicals.com. Archivado desde el original el 7 de julio de 2011. Consultado el 12 de mayo de 2009.
- «AM Chemicals, LLC, a supplier of solid supports and reagents for oligonucleotide and organic synthesis on solid phase». Amchemicals.com. Archivado desde el original el 7 de julio de 2011. Consultado el 12 de mayo de 2009.
- Powell, M. (17 de enero de 2008). «Supports». Glenresearch.com. Archivado desde el original el 6 de enero de 2009. Consultado el 12 de mayo de 2009.
- Azhayev, A. V.; Antopolsky, M. L. (2001). «Amide group assisted 3′-dephosphorylation of oligonucleotides synthesized on universal A-supports». Tetrahedron 57 (23): 4977-4986. doi:10.1016/S0040-4020(01)00409-4.
- «Metkinen Universal Solid Support III». Metkinenchemistry.com. Consultado el 4 de abril de 2012.
- «Glen Research Corporation products for DNA and RNA oligo synthesis – Support – 27-5010, Universal Support III PS». Glenresearch.com. 14 de noviembre de 2008. Archivado desde el original el 2 de diciembre de 2008. Consultado el 12 de mayo de 2009.
- «Glen Research Report of Products for RNA and DNA Oligonucelotide Synthesis, Modification and Labelling». Glenres.com. 17 de enero de 2008. Consultado el 12 de mayo de 2009.
- Petrie, C. R.; Reed, M. W.; Adams, A. D.; Meyer Jr, R. B. (1992). «An improved CPG support for the synthesis of 3'-amine-tailed oligonucleotides». Bioconjugate Chem. 3 (1): 85-87. PMID 1616954. doi:10.1021/bc00013a014.
- Lebedev, A. V.; Wickstrom, E. (1996). «The chirality problem in P-substituted oligonucleotides». Perspectives in Drug Discovery and Design 4 (1): 17-40. doi:10.1007/BF02172106.
- Wilk, A.; Grajkowski, A.; Phillips, L. R.; Beaucage, S. L. (2000). «Deoxyribonucleoside Cyclic N-Acylphosphoramidites as a New Class of Monomers for the Stereocontrolled Synthesis of Oligothymidylyl- and Oligodeoxycytidylyl- Phosphorothioates». J. Amer. Chem. Soc. 122 (10): 2149-2156. doi:10.1021/ja991773u.
- «Glen Research Report of Products for RNA and DNA Oligonucelotide Synthesis, Modification and Labelling». Glenresearch.com. 17 de enero de 2008. Consultado el 12 de mayo de 2009.
- «Sulfurizing reagent ii and its use in synthesizing oligonucleotide phosphorothioates». Glen research 18 (1). 2006. Archivado desde el original el 2 de diciembre de 2008. Consultado el 1 de agosto de 2009.
- «AM Chemicals, LLC, a supplier of solid supports and reagents for oligonucleotide and organic synthesis on solid phase». Amchemicals.com. Archivado desde el original el 18 de febrero de 2009. Consultado el 12 de mayo de 2009.
- «Glen Research Corporation products for DNA and RNA oligo synthesis – Minor Base – 40-4037, Sulfurizing Reagent II». Glenresearch.com. 14 de noviembre de 2008. Archivado desde el original el 11 de julio de 2011. Consultado el 12 de mayo de 2009.
- Iyer, R. P.; Egan, W.; Regan, J. B.; Beaucage, S. L. (1990). «3H-1,2-Benzodithiole-3-one 1,1-dioxide as an improved sulfurizing reagent in the solid-phase synthesis of oligodeoxyribonucleoside phosphorothioates». J. Amer. Chem. Soc. 112 (3): 1253-1254. doi:10.1021/ja00159a059.
- Beaucage, S. L. (2001). «3H-1,2-benzodithiol-3-one 1,1-dioxide». e-EROS Encyclopedia of Reagents for Organic Synthesis. doi:10.1002/047084289X.rn00167.
- «3400/394/392/391 DNA Synthesizer Reagents». Products.appliedbiosystems.com. Consultado el 12 de mayo de 2009.
- Vu, H.; Hirschbein, B. L. (1991). «Internucleotide phosphite sulfurization with tetraethylthiuram disulfide. Phosphorothioate oligonucleotide synthesis via phosphoramidite chemistry». Tetrahedron Lett. 32 (26): 3005-3008. doi:10.1016/0040-4039(91)80672-S.
- Tanaka, Toshiki; Letsinger, R. L. (1982). «Syringe method for stepwise chemical synthesis of oligonucleotides». Nucl. Acids Res. 10 (10): 3249-3259. doi:10.1093/nar/10.10.3249.
- «OligoMaster LS2». Azcobiotech.com. Archivado desde el original el 10 de noviembre de 2011. Consultado el 18 de octubre de 2011.
- «DNA / RNA Oligonucleotide Synthesizer: MerMade 384». Bioautomation.com. Archivado desde el original el 30 de septiembre de 2011. Consultado el 18 de octubre de 2011.
- «DNA / RNA Oligonucleotide Synthesizer: MerMade». Bioautomation.com. Consultado el 18 de octubre de 2011.
- «Azco DNA/RNA Synthesizers». Azcobiotech.com. Archivado desde el original el 15 de septiembre de 2011. Consultado el 18 de octubre de 2011.
- «Instruments». Biosset.com. Archivado desde el original el 3 de noviembre de 2012. Consultado el 4 de abril de 2012.
- «3900 High-Throughput DNA Synthesizer and Accessories». Products.appliedbiosystems.com. 28 de marzo de 2008. Consultado el 18 de octubre de 2011.
- «Polygen GmbH – Experiences & know-how in development & manufacturing DNA synthesizers». Polygen.de. Consultado el 18 de octubre de 2011.
- «GE Healthcare Life Sciences – Products – Oligonucleotide Synthesizers». Gelifesciences.com. Archivado desde el original el 7 de agosto de 2011. Consultado el 18 de octubre de 2011.
- «QMaster DNA/RNA Synthesizer». Genomictechnologies.com.
- «QMaster DNA/RNA Synthesizer». www.genomictechnologies.com/QmasterII.shtml. Archivado desde el original el 4 de marzo de 2016. Consultado el 2 de junio de 2016.
- Sanghvi, Y. S. (2011). «A status update of modified oligonucleotides for chemoterapeutics applications». Curr. Protoc. Nucleic Acid Chem. 46 (16): 4.1.1-4.1.22. doi:10.1002/0471142700.nc0401s46.
- Pease A. C., Solas D., Sullivan E. J., Cronin M. T., Holmes C.P., Fodor S. P. (1994). «Light-generated oligonucleotide arrays for rapid DNA sequence analysis». Proc. Natl. Acad. Sci. U.S.A. 91 (11): 5022-5026. Bibcode:1994PNAS...91.5022P. PMC 43922. PMID 8197176. doi:10.1073/pnas.91.11.5022.
- Egeland, R. D; Southern, E. M. (2005). «Electrochemically directed synthesis of oligonucleotides for DNA microarray fabrication» (Free full text). Nucl. Acids Res. 33 (14): e125. PMC 1183109. PMID 16085751. doi:10.1093/nar/gni117.
- Capaldi, D. C.; Gaus, H.; Krotz, A. H. (2003). «Synthesis of High-Quality Antisense Drugs. Addition of Acrylonitrile to Phosphorothioate Oligonucleotides: Adduct Characterization and Avoidance». Org. Proc. Res. & Development 7 (6): 832-838. doi:10.1021/op020090n.
- Volume 5: Deprotect to completion in organic solvents. Glen Report 22 (2)
- Boal, J. H.; Wilk, A.; Harindranath, N.; Max, E. E.; Kempel, T.; Beaucage, S. L. (1996). «Cleavage of oligodeoxyribonucleotides from controlled-pore glass supports and their rapid deprotection by gaseous amines». Nucl. Acids Res. 24 (15): 3115-7. PMC 146024. PMID 8760903. doi:10.1093/nar/24.15.3115.
- Westman, E.; Stroemberg, R. (1994). «Removal of t-butyldimethylsilyl protection in RNA-synthesis. Triethylamine trihydrofluoride (TEA, 3HF) is a more reliable alternative to tetrabutylammonium fluoride (TBAF)». Nucl. Acids Res. 22 (12): 2430. doi:10.1093/nar/22.12.2430.
- Krotz, A. H; Gaus, H.; Hardee, G. E. (2005). «Formation of oligonucleotide adducts in pharmaceutical formulations». Pharmaceutical development and technology 10 (2): 283-90. PMID 15926677. doi:10.1081/PDT-54464.
- Willems, A.; Deforce, D. L.; Van Bocxlaer, J. (2008). «Analysis of oligonucleotides using capillary zone electrophoresis and electrospray mass spectrometry, in Methods in Molecular Biology». Capillary Electrophoresis 384 (Totowa, NJ). pp. 401-414. doi:10.1007/978-1-59745-376-9_14.
Más lecturas
- Comprehensive Natural Products Chemistry, Volume 7: DNA and Aspects of Molecular Biology. Kool, Eric T., Editor. Neth. (1999), 733 pp. Publisher: (Elsevier, Ámsterdam, Neth.)
- Beaucage, S. L.; Iyer, R. P. (1992). «Advances in the synthesis of oligonucleotides by the phosphoramidite approach». Tetrahedron 48: 2223-2311. doi:10.1016/s0040-4020(01)88752-4.
- Beaucage, S. L.; Iyer, R. P. (1993). «The functionalization of oligonucleotides via phosphoramidite derivatives». Tetrahedron 49: 1925-1963. doi:10.1016/s0040-4020(01)86295-5.
- Beaucage, S. L.; Iyer, R. P. (1993). «The synthesis of modified oligonucleotides by the phosphoramidite approach and their applications». Tetrahedron 49: 6123-6194. doi:10.1016/s0040-4020(01)87958-8.
- Beaucage, S L. "Oligodeoxyribonucleotides synthesis. Phosphoramidite approach. Methods in Molecular Biology (Totowa, NJ, United States) (1993), 20 (Protocols for Oligonucleotides and Analogs), 33–61.
- Reese, C. B. (2002). «The chemical synthesis of oligo- and poly-nucleotides: a personal commentary». Tetrahedron 58: 8893-8920. doi:10.1016/s0040-4020(02)01084-0.
- Glaser, Vicki (1 de mayo de 2009). «Oligo Market Benefits from RNAi Focus». Genetic Engineering & Biotechnology News. Bioprocessing 29 (9) (Mary Ann Liebert). pp. 46-49. ISSN 1935-472X. OCLC 77706455. Archivado desde el original el 16 de abril de 2010. Consultado el 25 de julio de 2009.
Enlaces externos
Véase también
- Ácidos nucleicos
- Análogos de ácidos nucleicos
- Péptido de ácido nucleico
| Control de autoridades |
|
|---|
 Datos: Q2620256
Datos: Q2620256