Secuenciación de escopeta
En genética, la secuenciación de escopeta o shotgun sequencing es un método utilizado para secuenciar cadenas de ADN aleatorias. Recibe su nombre por analogía con el patrón de disparo cuasialeatorio de rápida expansión de una escopeta.
El método de secuenciación de ADN por terminación de cadena ("secuenciación de Sanger") solo se puede utilizar para cadenas cortas de ADN de 100 a 1000 pares de bases. Debido a este límite de tamaño, las secuencias más largas se subdividen en fragmentos más pequeños que se pueden secuenciar por separado, y estas secuencias se ensamblan para dar la secuencia general.
Hay dos métodos principales para este proceso de fragmentación y secuenciación. Primer walking (o "chromosome walking") progresa a través de toda la hebra pieza por pieza, mientras que la secuenciación rápida es un proceso más rápido, pero más complejo que utiliza fragmentos aleatorios.
En la secuenciación de escopeta,[1][2] ADN se divide al azar en numerosos segmentos pequeños, que se secuencian utilizando el método de terminación de cadena para obtener lecturas. Se obtienen múltiples lecturas superpuestas para el ADN diana realizando varias rondas de esta fragmentación y secuenciación. Los programas de computadora luego usan los extremos superpuestos de diferentes lecturas para ensamblarlos en una secuencia continua.
La secuenciación por escopeta fue una de las tecnologías precursoras que fue responsable de permitir la secuenciación completa del genoma.
Ejemplo
Por ejemplo, considere las siguientes dos rondas de lecturas de escopeta:
| Hebra | Secuencia |
|---|---|
| Original | AGCATGCTGCAGTCATGCTTAGGCTA |
| Primera secuencia de escopeta | AGCATGCTGCAGTCATGCT--------------------------TAGGCTA |
| Segunda secuencia de escopeta | AGCATG--------------------------CTGCAGTCATGCTTAGGCTA |
| Reconstrucción | AGCATGCTGCAGTCATGCTTAGGCTA |
En este ejemplo extremadamente simplificado, ninguna de las lecturas cubre la longitud completa de la secuencia original, pero las cuatro lecturas se pueden ensamblar en la secuencia original usando la superposición de sus extremos para alinearlas y ordenarlas. En realidad, este proceso utiliza enormes cantidades de información plagada de ambigüedades y errores de secuenciación. El ensamblaje de genomas complejos también se complica por la gran abundancia de secuencias repetitivas, lo que significa que lecturas cortas similares podrían provenir de partes completamente diferentes de la secuencia.
Se necesitan muchas lecturas superpuestas para cada segmento del ADN original para superar estas dificultades y ensamblar con precisión la secuencia. Por ejemplo, para completar el Proyecto Genoma Humano, la mayor parte del genoma humano se secuenció a una cobertura 12X o mayor; es decir, cada base de la secuencia final estuvo presente en promedio en 12 lecturas diferentes. Aun así, los métodos actuales no han logrado aislar o ensamblar una secuencia confiable para aproximadamente el 1% del genoma humano (eucromático), hasta 2004.[3]
Secuenciación escopeta del genoma completo
Historia
La secuenciación rápida del genoma completo para genomas pequeños (4000 a 7000 pares de bases) se sugirió por primera vez en 1979.[1] El primer genoma secuenciado por secuenciación de escopeta fue el del virus del mosaico de la coliflor, publicado en 1981.[4][5]
Secuenciación paired-end
La aplicación más amplia se benefició de la secuenciación de extremos por pares, conocida coloquialmente como secuenciación de escopeta de doble cañón (double-barrel shotgun sequencing). A medida que los proyectos de secuenciación comenzaron a asumir secuencias de ADN más largas y complicadas, varios grupos comenzaron a darse cuenta de que se podía obtener información útil secuenciando ambos extremos de un fragmento de ADN. Aunque secuenciar ambos extremos del mismo fragmento y realizar un seguimiento de los datos emparejados fue más engorroso que secuenciar un solo extremo de dos fragmentos distintos, saber que las dos secuencias estaban orientadas en direcciones opuestas y tenían aproximadamente la longitud de un fragmento aparte de cada uno fue valioso para reconstruir la secuencia del fragmento objetivo original.
Historia. La primera descripción publicada del uso de extremos emparejados fue en 1990[6] como parte de la secuenciación del locus HGPRT humano, aunque el uso de extremos emparejados se limitó a cerrar brechas después de la aplicación de un enfoque de secuenciación de escopeta tradicional. La primera descripción teórica de una estrategia de secuenciación de extremos por pares pura, asumiendo fragmentos de longitud constante, fue en 1991.[7] En ese momento, existía un consenso de la comunidad de que la longitud óptima del fragmento para la secuenciación final por pares sería tres veces la longitud de lectura de la secuencia. En 1995, Roach et al.[8] introdujo la innovación de usar fragmentos de diferentes tamaños y demostró que una estrategia pura de secuenciación final por pares sería posible en grandes objetivos. La estrategia fue posteriormente adoptada por el Instituto de Investigación Genómica (TIGR) para secuenciar el genoma de la bacteria Haemophilus influenzae en 1995,[9] y luego por Celera Genomics para secuenciar el genoma de Drosophila melanogaster (mosca de la fruta) en 2000,[10] y posteriormente el genoma humano.
Acercamiento
Para aplicar la estrategia, una hebra de ADN de alto peso molecular se corta en fragmentos aleatorios, se selecciona por tamaño (generalmente 2, 10, 50 y 150 kb) y se clona en un vector apropiado. A continuación, los clones se secuencian desde ambos extremos usando el método de terminación de cadena produciendo dos secuencias cortas. Cada secuencia se denomina lectura final o lectura 1 y lectura 2 y dos lecturas del mismo clon se denominan pares de apareamiento. Dado que el método de terminación de la cadena generalmente solo puede producir lecturas de entre 500 y 1000 bases de largo, en todos los clones, excepto en los más pequeños, los pares de apareamiento rara vez se superponen.
Montaje
La secuencia original se reconstruye a partir de las lecturas utilizando un software de ensamblaje de secuencias. Primero, las lecturas superpuestas se recopilan en secuencias compuestas más largas conocidas como contigs. Los contigs se pueden unir en andamios siguiendo las conexiones entre pares de aparejos. La distancia entre los contigs se puede inferir de las posiciones de los pares coincidentes si se conoce la longitud promedio del fragmento de la biblioteca y tiene una ventana de desviación estrecha. Dependiendo del tamaño del espacio entre contigs, se pueden usar diferentes técnicas para encontrar la secuencia en los espacios. Si el espacio es pequeño (5-20 kb), entonces se requiere el uso de la reacción en cadena de la polimerasa (PCR) para amplificar la región, seguida de la secuenciación. Si la brecha es grande (>20 kb), el fragmento grande se clona en vectores especiales como los cromosomas artificiales bacterianos (BAC) seguido de la secuenciación del vector.
Pros y contras
Los defensores de este enfoque argumentan que es posible secuenciar todo el genoma a la vez utilizando grandes conjuntos de secuenciadores, lo que hace que todo el proceso sea mucho más eficiente que los enfoques más tradicionales. Los detractores argumentan que, aunque la técnica secuencia rápidamente grandes regiones de ADN, su capacidad para unir correctamente estas regiones es sospechosa, particularmente para genomas con regiones repetidas. A medida que los programas de ensamblaje de secuencias se vuelven más sofisticados y la potencia informática se vuelve más barata, puede ser posible superar esta limitación.
Cobertura
La cobertura (profundidad o profundidad de lectura) es el número medio de lecturas que representan un nucleótido dado en la secuencia reconstruida. Se puede calcular a partir de la longitud del genoma original (G), el número de lecturas (N) y la longitud media de lectura (L) como . Por ejemplo, un genoma hipotético con 2.000 pares de bases reconstruidos a partir de 8 lecturas con una longitud promedio de 500 nucleótidos tendrá una redundancia 2x. Este parámetro también permite estimar otras cantidades, como el porcentaje del genoma cubierto por lecturas (a veces también llamado cobertura). Se desea una alta cobertura en la secuenciación de escopeta porque puede superar errores en la llamada y el ensamblaje de bases. El tema de la teoría de la secuenciación del ADN aborda las relaciones de tales cantidades.

A veces se hace una distinción entre cobertura de secuencia y cobertura física. La cobertura de secuencia es el número promedio de veces que se lee una base. La cobertura física es el número medio de veces que se lee o se amplía una base mediante lecturas emparejadas.[11]
Secuencia de escopeta jerárquica
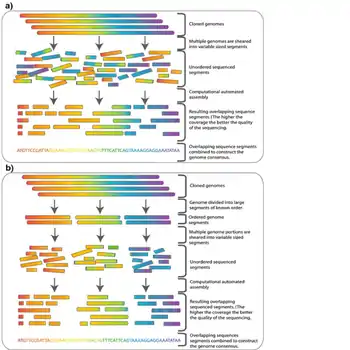
Aunque, en teoría, la secuenciación rápida se puede aplicar a un genoma de cualquier tamaño, su aplicación directa a la secuenciación de genomas grandes (por ejemplo, el genoma humano) fue limitada hasta finales de la década de 1990, cuando los avances tecnológicos hicieron práctico el manejo de grandes cantidades de datos complejos involucrados en el proceso.[12] Históricamente, se creía que la secuenciación de escopeta de genoma completo estaba limitada tanto por el gran tamaño de los genomas grandes como por la complejidad agregada por el alto porcentaje de ADN repetitivo (más del 50% para el genoma humano) presente en genomas grandes.[13] No fue ampliamente aceptado que una secuencia de escopeta de genoma completo de un genoma grande proporcionaría datos confiables. Por estas razones, se tuvieron que utilizar otras estrategias que redujeron la carga computacional del ensamblaje de secuencias antes de realizar la secuenciación de escopeta. En la secuenciación jerárquica, también conocida como secuenciación de arriba hacia abajo, se realiza un mapa físico de baja resolución del genoma antes de la secuenciación real. A partir de este mapa, se selecciona un número mínimo de fragmentos que cubren todo el cromosoma para secuenciar.[14] De esta manera, se requiere la cantidad mínima de secuenciación y ensamblaje de alto rendimiento.
El genoma amplificado primero se corta en trozos más grandes (50-200 kb) y se clona en un huésped bacteriano utilizando BAC o cromosomas artificiales derivados de P1 (PAC). Debido a que se han cortado al azar múltiples copias del genoma, los fragmentos contenidos en estos clones tienen diferentes extremos, y con suficiente cobertura encontrar un andamio de contigs BAC que cubra todo el genoma es teóricamente posible. Este andamio se llama camino de mosaico (tiling path).
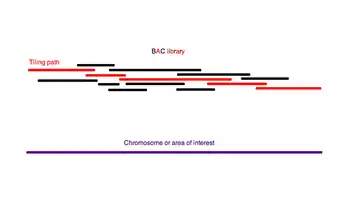
Una vez que se ha encontrado una ruta de mosaico, los BAC que forman esta ruta se cortan al azar en fragmentos más pequeños y se pueden secuenciar utilizando el método de la escopeta en una escala más pequeña.
Aunque no se conocen las secuencias completas de los contigs BAC, se conocen sus orientaciones entre sí. Existen varios métodos para deducir este orden y seleccionar los BAC que componen una ruta de mosaico. La estrategia general implica identificar las posiciones de los clones entre sí y luego seleccionar la menor cantidad de clones necesarios para formar un andamio contiguo que cubra toda el área de interés. El orden de los clones se deduce determinando la forma en que se superponen.[15] Los clones superpuestos se pueden identificar de varias formas. Una pequeña sonda marcada radioactivamente o químicamente que contiene un sitio marcado con secuencia (STS) se puede hibridar en una micromatriz sobre la que se imprimen los clones. De esta forma, se identifican todos los clones que contienen una secuencia particular en el genoma. El final de uno de estos clones se puede secuenciar para producir una nueva sonda y el proceso se repite en un método llamado caminata cromosómica.
Alternativamente, la biblioteca BAC se puede digerir por restricción. Se infiere que dos clones que tienen varios tamaños de fragmentos en común se superponen porque contienen en común múltiples sitios de restricción espaciados de manera similar.[15] Este método de mapeo genómico se denomina huella dactilar de restricción porque identifica un conjunto de sitios de restricción contenidos en cada clon. Una vez que se ha encontrado la superposición entre los clones y se conoce su orden en relación con el genoma, se secuencia como escopeta un andamio de un subconjunto mínimo de estos contigs que cubre todo el genoma.[14]
Debido a que primero implica la creación de un mapa de baja resolución del genoma, la secuenciación jerárquica de escopeta es más lenta que la secuenciación de escopeta de genoma completo, pero depende menos de los algoritmos informáticos que la secuenciación de escopeta de genoma completo. Sin embargo, el proceso de creación extensiva de bibliotecas BAC y selección de rutas en mosaico hace que la secuenciación de escopeta jerárquica sea lenta y laboriosa. Ahora que la tecnología está disponible y se ha demostrado la fiabilidad de los datos,[13] la velocidad y la rentabilidad de la secuenciación rápida del genoma completo lo han convertido en el método principal para la secuenciación del genoma.
Nuevas tecnologías de secuenciación
La secuenciación de escopeta clásica se basó en el método de secuenciación de Sanger: esta fue la técnica más avanzada para secuenciar genomas desde aproximadamente 1995-2005. La estrategia de la escopeta todavía se aplica hoy, sin embargo, se utilizan otras tecnologías de secuenciación, como la secuenciación de lectura corta y la secuenciación de lectura larga.
La secuenciación de lectura corta o de “próxima generación” produce lecturas más cortas (entre 25 y 500 pb) pero muchos cientos de miles o millones de lecturas en un tiempo relativamente corto (del orden de un día).[16] Esto da como resultado una alta cobertura, pero el proceso de ensamblaje es mucho más intensivo en computación. Estas tecnologías son muy superiores a la secuenciación de Sanger debido al gran volumen de datos y al tiempo relativamente corto que lleva secuenciar un genoma completo.[17]
Secuenciación de escopeta metagenómica
Tener lecturas de 400-500 pares de bases de longitud es suficiente para determinar la especie/cepa del organismo de donde proviene el ADN, siempre que su genoma ya sea conocido, utilizando por ejemplo un software clasificador taxonómico basado en k-meros. Con millones de lecturas de la secuenciación de próxima generación de una muestra ambiental, es posible obtener una descripción completa de cualquier microbioma complejo con miles de especies, como la flora intestinal. Las ventajas sobre la secuenciación de amplicones de ARNr 16S son: no limitarse a bacterias; clasificación a nivel de cepa donde la secuenciación de amplicones solo obtiene el género; y la posibilidad de extraer genes completos y especificar su función como parte del metagenoma.[18] La sensibilidad de la secuenciación metagenómica la convierte en una opción atractiva para uso clínico.[19] Sin embargo, enfatiza el problema de la contaminación de la muestra o la tubería de secuenciación.[20]
Véase también
- Teoría de la secuenciación del ADN
- Secuenciación clínica metagenómica
Referencias
- Staden, R (1979). «A strategy of DNA sequencing employing computer programs». Nucleic Acids Research 6 (70): 2601-10. PMC 327874. PMID 461197. doi:10.1093/nar/6.7.2601.
- Anderson, Stephen (10 de julio de 1981). «Shotgun DNA sequencing using cloned DNase I-generated fragments». Nucleic Acids Research (en inglés) 9 (13): 3015-3027. ISSN 0305-1048. PMC 327328. PMID 6269069. doi:10.1093/nar/9.13.3015.
- International Human Genome Sequencing Consortium (21 de octubre de 2004). «Finishing the euchromatic sequence of the human genome». Nature 431 (7011): 931-945. ISSN 1476-4687. PMID 15496913. doi:10.1038/nature03001.
- Gardner, R C; Howarth, A J; Hahn, P; Brown-Luedi, M; Shepherd, R J; Messing, J (25 de junio de 1981). «The complete nucleotide sequence of an infectious clone of cauliflower mosaic virus by M13mp7 shotgun sequencing.». Nucleic Acids Research 9 (12): 2871-2888. ISSN 0305-1048. PMID 6269062.
- Doctrow, Brian (19 de julio de 2016). «Profile of Joachim Messing». Proceedings of the National Academy of Sciences of the United States of America 113 (29): 7935-7937. ISSN 0027-8424. PMC 4961156. PMID 27382176. doi:10.1073/pnas.1608857113.
- Edwards, Al; Caskey, C. Thomas (1 de agosto de 1991). «Closure strategies for random DNA sequencing». Methods (en inglés) 3 (1): 41-47. ISSN 1046-2023. doi:10.1016/S1046-2023(05)80162-8.
- Edwards, A.; Voss, H.; Rice, P.; Civitello, A.; Stegemann, J.; Schwager, C.; Zimmermann, J.; Erfle, H. et al. (1990-04). «Automated DNA sequencing of the human HPRT locus». Genomics 6 (4): 593-608. ISSN 0888-7543. PMID 2341149. doi:10.1016/0888-7543(90)90493-e.
- Roach, J. C.; Boysen, C.; Wang, K.; Hood, L. (20 de marzo de 1995). «Pairwise end sequencing: a unified approach to genomic mapping and sequencing». Genomics 26 (2): 345-353. ISSN 0888-7543. PMID 7601461. doi:10.1016/0888-7543(95)80219-c.
- Fleischmann, R. D.; Adams, M. D.; White, O.; Clayton, R. A.; Kirkness, E. F.; Kerlavage, A. R.; Bult, C. J.; Tomb, J. F. et al. (28 de julio de 1995). «Whole-genome random sequencing and assembly of Haemophilus influenzae Rd». Science (New York, N.Y.) 269 (5223): 496-512. ISSN 0036-8075. PMID 7542800. doi:10.1126/science.7542800.
- Adams, M. D.; Celniker, S. E.; Holt, R. A.; Evans, C. A.; Gocayne, J. D.; Amanatides, P. G.; Scherer, S. E.; Li, P. W. et al. (24 de marzo de 2000). «The genome sequence of Drosophila melanogaster». Science (New York, N.Y.) 287 (5461): 2185-2195. ISSN 0036-8075. PMID 10731132. doi:10.1126/science.287.5461.2185.
- Meyerson, Matthew; Gabriel, Stacey; Getz, Gad (2010-10). «Advances in understanding cancer genomes through second-generation sequencing». Nature Reviews. Genetics 11 (10): 685-696. ISSN 1471-0064. PMID 20847746. doi:10.1038/nrg2841.
- Dunham, Ian (2005). eLS (en inglés). American Cancer Society. ISBN 978-0-470-01590-2. doi:10.1038/npg.els.0005378.
- Venter, J. C. ‘’Shotgunning the Human Genome: A Personal View.’’ Encyclopedia of Life Sciences, 2006.
- Gibson, G. and Muse, S. V. A Primer of Genome Science. 3rd ed. P.84
- Dear, P. H. Genome Mapping. Encyclopedia of Life Sciences, 2005. doi 10.1038/npg.els.0005353.
- Voelkerding, Karl V.; Dames, Shale A.; Durtschi, Jacob D. (2009-04). «Next-generation sequencing: from basic research to diagnostics». Clinical Chemistry 55 (4): 641-658. ISSN 1530-8561. PMID 19246620. doi:10.1373/clinchem.2008.112789.
- Metzker, Michael L. (2010-01). «Sequencing technologies - the next generation». Nature Reviews. Genetics 11 (1): 31-46. ISSN 1471-0064. PMID 19997069. doi:10.1038/nrg2626.
- Roumpeka, Despoina D.; Wallace, R. John; Escalettes, Frank; Fotheringham, Ian; Watson, Mick (2017). «A Review of Bioinformatics Tools for Bio-Prospecting from Metagenomic Sequence Data». Frontiers in Genetics 8: 23. ISSN 1664-8021. PMC 5337752. PMID 28321234. doi:10.3389/fgene.2017.00023.
- Gu, Wei; Miller, Steve; Chiu, Charles Y. (24 de enero de 2019). «Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection». Annual review of pathology 14: 319-338. ISSN 1553-4006. PMC 6345613. PMID 30355154. doi:10.1146/annurev-pathmechdis-012418-012751.
- Thoendel, Matthew; Jeraldo, Patricio; Greenwood-Quaintance, Kerryl E.; Yao, Janet; Chia, Nicholas; Hanssen, Arlen D.; Abdel, Matthew P.; Patel, Robin (2017-6). «Impact of Contaminating DNA in Whole-Genome Amplification Kits Used for Metagenomic Shotgun Sequencing for Infection Diagnosis». Journal of Clinical Microbiology 55 (6): 1789-1801. ISSN 0095-1137. PMC 5442535. PMID 28356418. doi:10.1128/JCM.02402-16.
Otras lecturas
- «Shotgun sequencing comes of age». The Scientist. Archivado desde el original el 14 de mayo de 2011. Consultado el 31 de diciembre de 2002.
- «Shotgun sequencing finds nanoorganisms - Probe of acid mine drainage turns up unsuspected virus-sized Archaea». SpaceRef.com. Consultado el 23 de diciembre de 2006.
| Control de autoridades |
|
|---|
 Datos: Q1073526
Datos: Q1073526 Multimedia: Shotgun sequencing / Q1073526
Multimedia: Shotgun sequencing / Q1073526