Secuenciación de proteínas
La secuenciación de proteínas es el proceso práctico de determinar la secuencia de aminoácidos de toda o parte de una proteína o péptido. Esto sirve para identificar a la proteína o caracterizar sus modificaciones postraduccionales. Usualmente, la secuenciación parcial de una proteína provee la información suficiente (uno o más fragmentos) para identificarla con bases de datos de proteínas derivadas de la traducción conceptual de los genes.
Los dos métodos directos más importantes para secuenciar proteínas son la espectrometría de masas y la degradación de Edman [1] usando un secuenciador. Los métodos de espectrometría son actualmente los más utilizados para la secuenciación e identificación, sin embargo, la degradación de Edman aún se mantiene como una herramienta valiosa.
Determinación de la composición de aminoácidos
Es deseable conocer la composición desordenada de los aminoácidos previo a la determinación de la secuencia ordenada pues esto facilita el descubrimiento de errores en el proceso de secuenciación así como la distinción entre resultados ambiguos. El conocimiento de la frecuencia de cierto aminoácido puede ser usado también para elegir cual peptidasa usar para la digestión(separación) de la proteína. La incorporación errónea a las proteínas de bajos niveles de aminoácidos no estándar (como la norleucina) también puede ser determinada.[2] El método generalizado (usualmente nombrado como análisis de aminoácidos[3]) para determinar la frecuencia de aminoácidos es el siguiente:
- Hidrolizar una cantidad conocida de proteína en cada uno de los aminoácidos que la constituyen (residuos).
- Separar los residuos y cuantificar los aminoácidos.
- Obtener la frecuencia relativa al dividir las cantidades (mol) de aminoácido por el valor más pequeño encontrado.
Hidrólisis
La hidrólisis se realiza al calentar una muestra de proteína con ácido clorhídrico 6 mol L-1 a 100-110 °C durante 24 horas o más. Las proteínas con muchos grupos voluminosos e hidrofóbicos pueden requerir periodos de calentamiento mayores. Sin embargo, estas condiciones son tan fuertes que algunos aminoácidos (serina, treonina, tirosina, triptófano, glutamina y cisteína) se degradan. Para evitar este problema, se sugiere que se calienten muestras separadas durante diferentes tiempos, analizando cada disolución resultante y extrapolando a cero el tiempo de hidrólisis. También se sugiere una variedad de reactivos para prevenir o reducir la degradación como tioles o fenoles para proteger el triptófano y la tirosina del ataque del cloruro, así como pre-oxidar la cisteína. También se sugiere medir la cantidad de amoniaco formado para determinar la hidrólisis de las amidas.
Separación y cuantificación
Los aminoácidos pueden ser separados por cromatografía de intercambio iónico [4] y después derivatizados para facilitar su detección. Más comúnmente, los aminoácidos son derivatizados y después analizados por cromatografía de fase inversa.
Un ejemplo de la cromatografía de intercambio iónico se da por la NtrC (proteína C reguladora del nitrógeno) usando poliestireno sulfonatado como matriz (resina de intercambio iónico). Al añadir los aminoácidos en solución ácida y hacerlos pasar por un buffer que incremente poco a poco el pH, los aminoácidos son eluidos cuando el pH alcanza su respectivo punto isoeléctrico (pI). Como cada aminoácido posee un pI diferente, son separados. Una vez que los aminoácidos han sido separados, sus cantidades son determinadas al añadir un reactivo que formará un derivado colorido. Si las cantidades superan los 10 nmol, se puede utilizar ninhidrina, dando un color amarillo al reaccionar con la prolina y púrpura al reaccionar con el resto de los aminoácidos. La concentración de los aminoácidos es entonces proporcional a la absorbancia de la solución resultante (ley de Lambert-Beer) y se puede usar espectroscopía UV-visible. Con cantidades pequeñas (inferiores a los 10 pmol), se pueden formar derivados fluorescentes utilizando reactivos como ortoftaldehído o fluorescamina y se utiliza espectroscopía de fluorescencia.
La derivatización previa a la columna utiliza el reactivo de Edman para producir un derivado que es detectable por espectroscopía UV-visible. Una mayor sensibilidad es lograda al utilizar un reactivo que genere derivados fluorescentes. Los aminoácidos derivados son susceptibles de ser usados en cromatografía de fase inversa, usualmente usando una columna de gel de sílica C8 o C18 y un gradiente de elución optimizado. Los aminoácidos eluidos son detectados usando un detector de UV o de fluorescencia y los picos resultantes son comparados con estándares para cuantificar las muestras.
Análisis del aminoácido N-terminal
La determinación del aminoácido N-terminal de una cadena peptídica es útil por dos razones: para ayudar al ordenamiento de fragmentos en una cadena y porque el primer ciclo de la degradación de Edman usualmente se contamina por impurezas y no da una determinación certera del primer aminoácido N-terminal. Un método generalizado para el análisis del aminoácido N-terminal es:
- Hacer reaccionar el péptido con un reactivo que reaccione selectivamente con el aminoácido terminal.
- Hidrolizar la proteína.
- Determinar el aminoácido por cromatografía (u otros métodos) y comparar con los estándares.
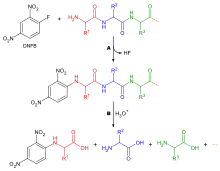
Existen diferentes reactivos que pueden ser usados para marcar el aminoácido terminal. Todos reaccionan con grupos amino y por lo tanto pueden enlazarse con aminas en las cadenas laterales como las de la lisina, por esta razón, es necesario ser cuidadoso en la interpretación de los cromatogramas para asegurar que se ha elegido el aminoácido correcto. Dos de los reactivos más comunes son el reactivo de Sanger (1-fluoro-2,4-dinitrobenceno) y derivados del dansilo. El fenilisotiocianato, reactivo usado en la degradación de Edman, también puede ser usado. Los reactivos producen derivados coloridos y solo se requiere análisis cualitativo por lo que los aminoácidos no necesitan ser eluidos de la columna de cromatografía, solo comparados con un estándar. Otra consideración es que, como la mayoría de los grupos amino reaccionan, la cromatografía de intercambio iónico no se puede usar. En su lugar, se usa cromatografía en capa fina y HPLC.
Análisis de aminoácido C-terminal
El número de métodos disponibles para el análisis del aminoácido C-terminal es mucho menor que para el aminoácido N-terminal. El método más común es añadir carboxipeptidasas a una solución de la proteína, tomar muestras en intervalos regulares y determinar el aminoácido terminal analizando una gráfica de concentraciones de aminoácido respecto al tiempo. Este método es muy útil en el caso de que los polipéptidos tengan el grupo N-terminal bloqueado. La secuenciación de grupos C-terminales es de gran ayuda en la verificación de estructuras primarias de proteínas predichas por las secuencias de ADN y para detectar algún procesamiento postraduccional de productos genéticos de secuencias de codones conocidas.
Degradación de Edman
La degradación de Edman es una reacción importante para la secuenciación de proteínas debido a que permite descubrir la composición ordenada de los aminoácidos de una proteína. Los secuenciadores automáticos de Edman son muy utilizados y permiten secuencias péptidos de 50 aminoácidos de largo. El procedimiento para secuenciar proteínas mediante la degradación de Edman es el siguiente:
- Romper cualquier puente disulfuro en la proteína con un agente reductor como el 2-mercaptoetanol. Un grupo protector como el ácido yodoacético es necesario para prevenir que los enlaces se regeneren.
- Separar y purificar las cadenas individuales del complejo proteico si hay más de una.
- Determinar la composición de aminoácidos de cada cadena.
- Determinar los aminoácidos terminales de cada cadena.
- Degradar cada cadena en fragmentos de menos de 50 aminoácidos.
- Separar y purificar los fragmentos.
- Determinar la secuencia de cada fragmento.
- Repetir con un patrón diferente de separación.
- Construir la secuencia de toda la proteína.
Digestión en fragmentos peptídicos
Péptidos mayores a 50 aminoácidos no pueden ser secuenciados confiablemente por la degradación de Edman pues aparecen interferencias. Debido a esto, cadenas proteicas largas deben ser separadas en fragmentos pequeños que después pueden ser secuenciados individualmente. La digestión se puede hacer mediante endopeptidasas como la tripsina o la pepsina o mediante agentes químicos como el bromuro de cianógeno. Enzimas diferentes dan patrones de separación diferentes. Los traslapes entre fragmentos pueden ser usados para construir una secuencia general.
Reacción
El péptido a secuenciar es adsorbido en una superficie sólida. Un sustrato común es la fibra de vidrio cubierta con polibreno (un polímero catiónico). El reactivo de Edman, fenilisotiocianato, es añadido al péptido adsorbido junto con una solución buffer ligeramente básica de trimetilamina al 12%. Esto reacciona con el grupo amino del aminoácido N-terminal.
El aminoácido terminal puede ser desorbido selectivamente por la adición de un ácido anhidro. El derivado se isomeriza para dar una fenilhidantoína sustituida, la cual puede ser enjuagada e identificada mediante cromatografía, después, el ciclo se repite. La eficiencia de cada paso es del 98%, lo que permite que se identifiquen cerca de 50 aminoácidos antes de perder la confiabilidad.

Secuenciador de proteínas
Un secuenciador de proteínas[5] es una máquina que permite llevar a cabo la degradación de Edman de manera automática. Una muestra de la proteína o péptido se coloca en el matraz de reacción del secuenciados y la degradación de Edman se lleva a cabo. Cada ciclo libera y derivatiza un aminoácido N-terminal de la proteína que es después identificado mediante HPLC. El proceso de secuenciación se repite para todo el polipéptido hasta que la secuencia completa se establece o hasta que se cumple un número de ciclos programado.
Identificación mediante espectrometría de masas
La identificación de las proteínas es el proceso de asignar un nombre a una proteína de interés (POI, protein of interest en inglés) basado en su secuencia de aminoácidos. Usualmente solo una parte de la proteína requiere ser determinada experimentalmente para identificar la proteína al compararla con bases de datos de secuencias proteicas deducidas de secuencias de ADN. La caracterización posterior incluye la confirmación de los extremos N y C de la POI.
Digestión proteolítica
Un esquema general de la identificación de proteínas es el siguiente:
- La POI se aísla, típicamente mediante SDS-PAGE o cromatografía.
- La proteína asilada puede ser modificada químicamente para estabilizar la cisteína.
- La POI es digerida con una proteasa específica para generar péptidos. La tripsina, que separa selectivamente el lado C-terminal de lisina y arginina, es la proteasa más usada. Entre las ventajas están: la frecuencia de aparición de la lisina y la arginina, la alta especificidad de la enzima, la estabilidad de la enzima y la compatibilidad de los péptidos trípticos para la espectrometría de masas.
- Los péptidos deben ser desalinizados para remover contaminantes ionizables y después sujetos a espectrometría de masas MALDI-TOF. Las medidas directas de las masas de los péptidos pueden proveer información suficiente para identificar la proteína (huella peptídica) aunque una fragmentación posterior de los péptidos dentro del espectrómetro de masas usualmente es usado para conseguir información de las secuencias de péptidos. Alternativamente, los péptidos pueden ser desalinizados y separados por cromatografía de fase inversa e introducidos en un espectrómetro de masas mediante una fuente ESI. El método cromatografía líquida-ESI-espectrómetro de masas (LC-ESI-MS) para la identificación provee más información pero requiere más tiempo
- Dependiendo del tipo de espectrómetro de masas, la fragmentación de iones peptídicos puede ocurrir mediante diversos mecanismos como la disociación por colisiones (CID, collision-induced dissociation) o decaimiento post-fuente (PSD, post-source decay). En cada caso, el patrón de los fragmentos iónicos de un péptido provee información acerca de su secuencia
- La información incluye las masas medidas de los iones peptídicos y posteriormente, estos fragmentos son comparados contra valores calculados de la proteólisis conceptual (in-silico) y contra una base de datos. Una comparación exitosa será encontrada si su puntuación excede el umbral de los parámetros. Incluso si la proteína no está representada en la base de datos, algunas máquinas permiten la identificación basándose en la similitud con proteínas homólogas.
- Los paquetes de software usualmente generan un reporte que muestra la identidad de cada proteína, su rango de similitud y una medida de la fuerza relativa de la similitud al identificar diversas proteínas.
- Un diagrama de los péptidos encontrados en la secuencia es usada para mostrar el porcentaje de detección de la proteína. Cuando la POI es significativamente menor que la proteína de la base de datos, el diagrama puede sugerir que la POI es un fragmento de la proteína identificada.
Referencias
- «Secuenciación de Edman». ProteomePlus. 12 de octubre de 2011. Consultado el 19 de abril de 2021.
- G, Bogosian (5 de enero de 1989). «Biosynthesis and Incorporation Into Protein of Norleucine by Escherichia Coli». The Journal of biological chemistry (en inglés). Consultado el 2 de mayo de 2020.
- Alterman, Michail A.; Hunziker, Peter. (2011). Amino acid analysis : methods and protocols. Humana. ISBN 978-1-61779-445-2. OCLC 764699045. Consultado el 2 de mayo de 2020.
- «¿Cómo la cromatografía de intercambio iónico trabaja?». News-Medical.net (en inglés). 9 de agosto de 2016. Consultado el 19 de abril de 2021.
- Edman, P.; Begg, G. (1967). «A Protein Sequenator». European Journal of Biochemistry (en inglés) 1 (1): 80-91. ISSN 1432-1033. doi:10.1111/j.1432-1033.1967.tb00047.x. Consultado el 2 de mayo de 2020.
 Datos: Q3142557
Datos: Q3142557