Tirosina aminotransferasa
La tirosina aminotransferasa o tirosina transaminasa (EC 2.6.1.5) es una enzima que cataliza la conversión de tirosina a 4-hidroxifenilpiruvato.[1]

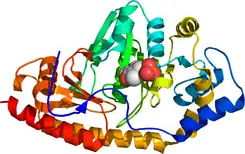
En humanos, la proteína tirosina aminotransferasa se encuentra codificada por el gen TAT. Una deficiencia de esta enzima en seres humanos produce la enfermedad conocida como tirosinemia tipo II, donde existe un exceso de tirosina en circulación como resultado de que la tirosina falla en convertirse en 4-hidroxifenilpiruvato.[2]
Clasificación
Esta enzima pertenece a la familia de las transferasas, más específicamente a aquellas transferasas que transfieren grupos amina (transaminasas).
Nomenclatura
El nombre sistemático de esta clase de enzimas es L-tirosina:2-oxoglutarato aminotransferasa. Otros nombres de uso común pueden ser:
- Tirosina transaminasa
- L-fenilalanina 2-oxoglutarato aminotransferasa
- L-tirosina aminotransferasa
- Glutámico fenilpirúvico aminotranferasa
- Glutámico-hidroxifenilpirúvico transaminasa
- Fenilalanina aminotransferasa
- Fenilalanina transaminasa
- Fenilalanina—α-cetoglutarato transaminasa
- Fenilpiruvato transaminasa
- Ácido fenilpirúvico transaminasa
- Tirosina aminotransferasa
- Tirosina—2-cetoglutarato aminotransferasa
- Tirosina—α-cetoglutarato aminotransferasa
- Tirosina—α-cetoglutarato transaminasa
- TyrAT
Mecanismo
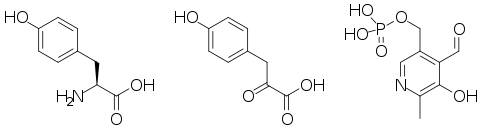
Un poco más abajo se muestran las estructuras de las tres principales moléculas involucradas en la reacción catalizada por la tirosna aminotransferasa: el aminoácido tirosina, el grupo prostético piridoxal fosfato, y el producto resultante 4-hidroxifenilpiruvato.
A cada lado de la proteína dimérica se encuentra un piridoxal fosfato (PLP) unido al residuo Lys280 de la molécula de tirosina aminotransferasa. El grupo amino de la tirosina ataca al carbono alfa de la imina unida a la Lys280, formando un complejo tetraédrico, y luego liberando la Lys-(Enzima). Este proceso se conoce como transiminación por acción del grupo imina unido al PLP. El nuevo complejo PLP-Tyr luego es atacado por una base.
Un posible candidato para el ataque de base en el mecanismo podría ser la Lys280 que acaba de ser expulsada del PLP, la cual secuestra el grupo amino recientemente formado en la unión PLP-Tyr. Por un mecanismo similar a la de la aspartato transaminasa, la lisina que forma inicialmente una imina con el PLP luego actúa como base que ataca a la tirosina en transiminación.
Los electrones que qudan detrás luego de la pérdida de un protón se mueven hacia abajo para formar un nuevo doble enlace con la imina, lo cual a su vez impulsa a los electrones que ya se encuentran formando un doble enlace a través del PLP terminando como un par solitario en el nitrógeno cargado positivamente del anillo de seis miembros que forma la molécula. Luego el carbono alfa de la imina PLP-Tyr es atacado por una molécula de agua y por medio de una sustitución de acilo expulsa al nitrógeno del PLP formando piridoxamina fosfato (PMP) y 4-hidroxifenilpiruvato.
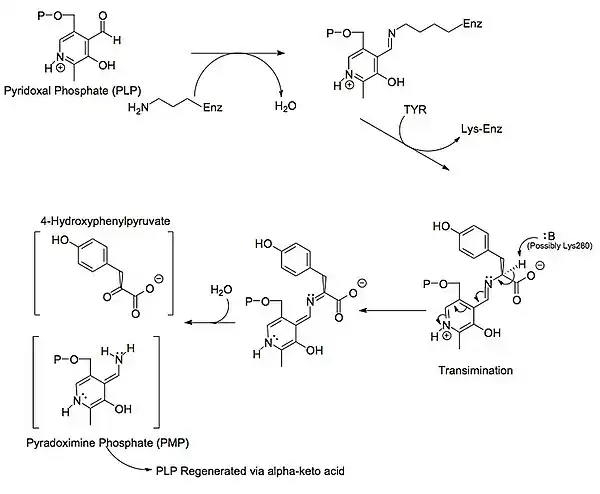
El PMP luego regenera al PLP por transferencia de su grupo amina al alfa-cetoglutarato, reformando su grupo aldehído funciona. Esto es seguido por otra reacción de sustitución en la que el residuo Lys280 regenera su enlace con la enzima formando nuevamente Enz-PLP.
Sitio activo
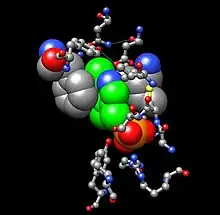
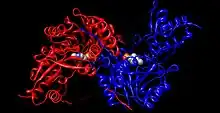
La tirosina aminotransferasa es una enzima dimérica con dos ranuras activas idénticas. La Lys280 se encuentra unida al PLP, el cual a su vez se encuentra sujeto en su sitio por dos cadenas laterales no polares; una fenilalanina y una isoleucina. Múltiples enlaces hidrógeno ayudan a mantener el PLP en posición, estos enlaces se producen principalmente en su grupo fosfato.
Abajo se muestra uno de los sitos activos en tres niveles diferentes de ampliación:

Patologías
La enfermedad metabólica más común asociada con la tirosina aminotransferasa es la tirosinemia. Esta enfermedad se produce como resultado de una deficiencia en la tirosina aminotransferasa hepática.[5] La tirosinemia tipo II (síndrome de Richner-Hanhart, RHS) es una enfermedad autosómica recesiva caracterizada por queratitis, hiperqueratosis palmoplantar, retraso mental y niveles elevados de tirosina en sangre.[5] La queratitis en los pacientes con tirosinemia tipo II es causada por el depósito de cristales de tirosina en la córnea, con la consiguiente inflamación corneal.[6]
El gen TAT en humanos se encuentra localizado en el brazo largo del cromosoma 16 (16q22-24) y se extiende sobre 10,9 kilobases, contiene 12 exones, y el ARNm que produce codifica para una proteína formada por 454 aminoácidos con un peso de 50,4 kDa.[7] Se han reportado doce diferentes mutaciones en el gen TAT.[7]
Referencias
- Dietrich JB (abril de 1992). «Tyrosine aminotransferase: a transaminase among others?». Cellular and molecular biology 38 (2): 95-114. PMID 1349265.
- Rettenmeier R, Natt E, Zentgraf H, Scherer G (julio de 1990). «Isolation and characterization of the human tyrosine aminotransferase gene». Nucleic Acids Res. 18 (13): 3853-61. PMC 331086. PMID 1973834. doi:10.1093/nar/18.13.3853.
- Pettersen, E.F., Goddard, T.D., Huang, C.C., Couch, G.S., Greenblatt, D.M., Meng, E.C., and Ferrin, T.E. (2004). «UCSF Chimera - A Visualization System for Exploratory Research and Analysis.». Comput. Chem. 25 (13): 1605-1612. PMID 15264254. doi:10.1002/jcc.20084.
- PDB 3DYD ; Karlberg T, Moche M, Andersson J, et al. (2008). «Human tyrosine aminotransferase». To be Published.
- Natt E, Kida K, Odievre M, Di Rocco M, Scherer G (octubre de 1992). «Point mutations in the tyrosine aminotransferase gene in tyrosinemia type II». Proc. Natl. Acad. Sci. U.S.A. 89 (19): 9297-301. PMC 50113. PMID 1357662. doi:10.1073/pnas.89.19.9297.
- al-Hemidan AI, al-Hazzaa SA (marzo de 1995). «Richner-Hanhart syndrome (tyrosinemia type II). Case report and literature review». Ophthalmic Genet. 16 (1): 21-6. PMID 7648039. doi:10.3109/13816819509057850.
- Minami-Hori M, Ishida-Yamamoto A, Katoh N, Takahashi H, Iizuka H (enero de 2006). «Richner-Hanhart syndrome: report of a case with a novel mutation of tyrosine aminotransferase». J. Dermatol. Sci. 41 (1): 82-4. PMID 16318910. doi:10.1016/j.jdermsci.2005.10.007.
Las imágenes de gráficos moleculares se crearon utilizando el paquete UCSF Chimera del Resource for Biocomputing, Visualization, and Informatics de la Universidad de California, San Franciso (supported by NIH P41 RR-01081).
Enlaces externos
- MeSH: Tyrosine+aminotransferase (en inglés)