Épidermolyse bulleuse simple
L'épidermolyse bulleuse simple est une classe des épidermolyses bulleuses héréditaires. Elle correspond à un groupe de maladies caractérisées par l’apparition de bulles après une sollicitation mécanique.
| Épidermolyse bulleuse épidermolytique | |
| Référence MIM | 131760-131800 131900-131960 601001 |
|---|---|
| Transmission | Dominante principalement |
| Chromosome | 17q12-q21-12q13 |
| Gène | KRT14-KRT5 |
| Empreinte parentale | Non |
| Mutation | Ponctuelle |
| Mutation de novo | Possible |
| Incidence | 1 sur 50000 naissances |
| Maladie génétiquement liée | Aucune |
| Diagnostic prénatal | Possible |
| Liste des maladies génétiques à gène identifié | |
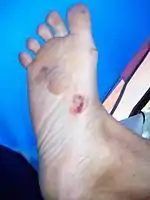
On regroupe sous le terme d’épidermolyse bulleuse l’ensemble des affections génétiques. Elles se caractérisent par la présence de lésions cutanées ou de bulles à la jonction derme-épiderme. Elle est observable à partir de la naissance ou au cours de l’enfance. La plupart des bulles se forment sur la lame basale de l’épiderme.
L'épidermolyse bulleuse simple, ou simplex, ou épidermolytique (EBS) est une maladie génétique. Elle se caractérise par une fragilité de la peau. Les connexions entre les différentes couches ne sont plus suffisantes. Elle se traduit par l'apparition de bulles ou de vésicules épidermiques. Celles-ci peuvent avoir plusieurs aspects. Elles se forment généralement à la suite d'un frottement (exemple : lors de la marche). Cette maladie est parfois appelée Maladie de Goldscheider (venant du neurologue Alfred Goldscheider). Il existe plusieurs formes d’épidermolyse bulleuse simple. Elles se différencient en fonction de l’âge de l’apparition de la maladie, de sa localisation et de la forme des «,bulles ».
Il s’agit d’une mutation à caractère dominante des gènes codants la kératine 5 ou 14. La kératine entre dans la composition des filaments intermédiaires des kératinocytes. Ces derniers sont à l’origine de cette maladie. Cela se localise au niveau des cellules épithéliales de la lame basale. L’épidermolyse bulleuse simple se retrouve sous des formes cliniques différentes : Koebner, Weber-Cockayne, Dowling-Meara, Ogna et épidermolyse bulleuse simple avec pigmentation mouchetée.
Les enfants atteints de cette maladie sont appelés les « enfants papillon ».
Types cliniques
L'EBS appartient au groupe des Épidermolyses bulleuses héréditaires (EBH). Les EBH sont des maladies génétiques cutanées, classées en trois groupes principaux : les EB simples, les EB dystrophiques et les EB jonctionnelles.
La recherche spécifique a découvert plus de 20 types différents d'EB dont cinq types cliniques du groupe simple :
- l’épidermolyse bulleuse simple de type Weber-Cockayne. Elle est aussi appelée épidermolyse bulleuse simple localisée. Ce type d’épidermolyse se présente avec des bulles uniquement au niveau des pieds et des mains. En effet, elle touche les paumes et les plantes. Les manifestations commencent rarement à la naissance. Les premiers signes apparaissent généralement vers 18 mois et parfois dans l'adolescence. Elle se révèle à la marche ou un peu plus tard. Les symptômes sont renforcés durant la période de l’été. Les traitements pour ce type d’épidermolyse bulleuse est le même que pour une hyperhidrose, c’est-à-dire une sudation excessive ;
- l'épidermolyse bulleuse simple de type Koebner, aussi appelée épidermolyse bulleuse simple généralisée. Les premières bulles apparaissent à la naissance ou dans la petite enfance. Les zones du corps atteintes sont nombreuses voir sur l'intégralité du corps dans les cas les plus graves ;
- l'épidermolyse bulleuse simple avec pigmentation mouchetée ou complètement dépigmentées. Lorsque la pigmentation mouchetée correspond à des taches brunes alternant avec des zones hypopigmentées (sous pigmentées). Alors que l‘épidermolyse bulleuse simple dépigmentée se situe surtout au niveau du tronc ou des mains. Ces troubles de la pigmentation disparaissent à l'âge adulte. Il s’agit des cas les plus rares et cette maladie peut causer des retards mentaux ;
- l'épidermolyse bulleuse herpétiforme type Dowling-Meara. Cette forme est plus sévère, parfois mortelle. Les bulles sont évidentes à la naissance ;
- l'épidermolyse bulleuse simple de type Ogna. Elle se caractérise par l'apparition de bulles hémorragiques au niveau des extrémités.
Diagnostic de la maladie
Parce que le clivage se fait très près de la jonction dermoépidermique, la microscopie électronique permet seule de faire la différence entre une EB dystrophique, jonctionnelle et simple. Pour préciser le plan de clivage, la microscopie électronique a été remplacée par l’immunohistochimie. Pour finir on réalise une immunofluorescence directe en peau saine congelée utilisant des anticorps dirigés contre la laminine 5, l'anti cytokératine 14, le collagène VII ou IV.
Le diagnostic du type Weber-Cockayne est presque toujours fait par la clinique. Les autres formes nécessitent la biopsie d'une bulle récente, l'étude au microscope électronique ou avec l'aide d'anticorps fluorescents met en évidence le type de lésions.
Le diagnostic génétique est possible mais rarement utilisé en pratique clinique. Il est surtout utilisé pour le conseil génétique[1].
Diagnostic différentiel :
Une dermatose bulleuse auto-immune est éliminée sur la négativité de l’IFD.
Origine et évolution de la maladie
L'origine est génétique. L'« accident » génétique provoque une défaillance du système d'adhésion des cellules dermiques et des différentes couches de l'épiderme.
La peau est rendue extrêmement fragile et se décolle au moindre frottement. De multiples petites vésicules agglutinées sont typiques de la maladie et la survenue de bulle hémorragique est habituelle.
La sévérité des manifestations varie grandement, tant dans une même famille qu'entre les individus touchés, allant de forme peu sévères à des formes mutilantes, voire mortelles. La douleur est permanente mais varie beaucoup selon le type d'EB.
Une hyperkératose des mains et des pieds survient dans l'enfance et peut devenir une source de plaintes majeures chez les personnes atteintes dans la vie adulte. Des troubles de la croissance des ongles et un millium cutané sont habituels. Une amélioration peut survenir tardivement dans l'enfance. La chaleur améliore l'état de certains malades.
Des troubles de l'alimentation traduisent une atteinte des muqueuses digestives.
Soins
En octobre et , une équipe de chercheurs et de médecins a réussi à soigner un patient atteint par cette maladie, en faisant une greffe de peau génétiquement modifiée[2],[3].
L’atteinte d’épidermolyse bulleuse chez les nouveau nés représente un risque infectieux majeur. La prise en charge pluridisciplinaire sera alors essentielle. Il sera nécessaire de mobiliser autant les infirmières, les dermatologues, une équipe de lutte contre la douleur, une diététicienne qu’une équipe de rééducation fonctionnelle. De plus, des spécialistes de la génétique permettent d’expliquer les raisons de la survenue de la maladie et même d’accompagner un couple dans un projet parental. Ce sont l’ensemble des interventions du personnel médical qui pourra optimiser un meilleur traitement de la maladie. Les soins de la peau sont souvent longs, soit plusieurs heures par jour, prolongés et douloureux. Ces soins nécessitent également d’être contrôlés régulièrement. De plus, il est important de savoir que la maladie d'épidermolyse bulleuse ne concerne pas seulement la peau. Elle peut entraîner des difficultés de déplacement, une atteinte oculaire, des difficultés alimentaires d’où le besoin d’être pris en charge par un corps médical pluridisciplinaire. Elle ne concerne donc pas que le côté visuel et le ressenti psychologique de la maladie.
Le diagnostic repose sur les informations cliniques et l'analyse immunohistochimique et ultrastructurale d'une biopsie de peau.
La prise en charge de cette pathologie comprend la protection de la peau contre les traumatismes et la prévention des infections secondaires.
L'activité des enfants doit diminuer les risques de traumatisme et les nouvelles bulles devront être percées. L'habillement se fera en trois couches : une non adhérente au contact de la peau, une deuxième protection rembourrée et la troisième élastique. L'utilisation du chlorure d'aluminium et du cyproheptadine peut réduire la formation de bulles chez certains individus.
L'utilisation de kératolytique et d'adoucissant de la peau permet de lutter contre l'hyperkératose.
Traitements
L'activité des enfants doit diminuer les risques de traumatisme et les nouvelles bulles devront être percées.
Le traitement a pour objectif d’éviter la formation de bulle en protégeant la peau, en évitant les traumatismes et en soignant les blessures. L'habillement se fera en trois couches : Une non adhérente au contact de la peau, une deuxième protection rembourrée pour la stabilité et la troisième élastique. L'utilisation du chlorure d'aluminium et du cyproheptadine peut réduire la formation de bulles chez certains individus.
L'utilisation de kératolytique et d'adoucissant de la peau permet de lutter contre l'hyperkératose.
Plusieurs situations sont à éviter. En effet, la chaleur peut exacerber les cloques et l’infection. Il est nécessaire de ne pas pratiquer d’activités qui traumatisent la peau. En ce qui concerne les pansements, il faut éviter le sparadrap classique.
Organismes à contacter
Il existe des établissements de référence en France pour les maladies rares telles que l'épidermolyse bulleuse. Ces centres ont été labellisés par le ministère de la santé, ils permettent d’améliorer le diagnostic et la prise en charge de ces maladies rares.
Quatre centres de référence :
- CHU de Nice ;
- Hôpital de Necker, Paris ;
- CHU de Bordeaux ;
- CHU de Toulouse (Centre de référence des maladies rares de la peau - Hôpital LARREY).
Six centres de compétence :
- Nantes ;
- Tours ;
- Saint-Étienne ;
- Dijon ;
- Lille ;
- Montpellier.
Dans ces centres, de nombreux experts aident les patients dans leur prise en charge tels que des dermatologues, généticiens, angiologues, chirurgiens, diététiciens, kinésithérapeutes, ergothérapeutes, ophtalmologistes, gastro-entérologues…
Mieux connaître sa maladie permettra d’améliorer la prise en charge du malade.
Notes et références
- Musette Ph., Prost-Squarcioni C., « Épidermolyse bulleuse acquise », sur www.therapeutique-dermatologique.org, (consulté le )
- France Inter, « La peau d'un enfant de 7 ans reconstituée à 80 % lors d'une greffe », sur France Inter, (consulté le )
- (en) Nature, « Skin regeneration with insights », sur Nature.com, (consulté le )
Voir aussi
Articles connexes
Bibliographie
- Dermatologie ; L.Valeyrie
- Dermatopathologie ; Springer, de W.Kempf, M.Hantschke, H.Kutzner, W.H.C Burgodf et R.G Panizzon
- Checklists de médecine, dermatologie, vénérologie, allergologie, phlébologie et andrologie de la collection Thieme Maloine. Les auteurs sont W.Sterry et R.Paus.
- Guide pratique de dermatologie de la collection Masson et écrit par Daniel Wallach.
- Histopathologie cutanée non tumorale de la collection sauramps medical. L’ouvrage a été coordonné par Janine Wechsler et un comité de rédaction : Sylvie Fraitag et Isabelle Moulonguet.
 Portail de la médecine
Portail de la médecine