Angiopathie amyloïde cérébrale
L'angiopathie amyloide cérébrale (abrégée en AAC) est une maladie cérébrovasculaire fréquente chez le sujet âgé, souvent asymptomatique et fortement associée avec la maladie d'Alzheimer. Elle résulte de dépôts amyloïdes (protéine de conformation anormale) dans la paroi des petits et moyens vaisseaux cérébraux ce qui les fragilise. Il s'agit en réalité d'une famille de maladies qui peuvent se différencier selon la composition du peptide amyloïde qui en est responsable. Cependant, épidémiologiquement c'est la forme secondaires aux dépôts de protéines Aβ-40 [une des formes de protéine β-amyloïde avec l'Aβ-42] qui domine. Les autres formes de la maladie sont très rares. La principale manifestation clinique de l'AAC est l'hémorragie intracérébrale dont elle est une cause fréquente (20 % des hémorragies cérébrales de l'adulte seraient attribuables à l'AAC).
-MRI.png.webp)
| Symptômes | Saignement (d) |
|---|
| Spécialité | Neurologie |
|---|
| CIM-10 | i68 |
|---|---|
| DiseasesDB | 32874 |
| MedlinePlus | 000719 |
| eMedicine | 1162720 |
| MeSH | D016657 |
![]() Mise en garde médicale
Mise en garde médicale
Épidémiologie
La prévalence exacte de l'AAC dans la population générale est mal connue car le diagnostic de certitude repose sur l'autopsie et la majorité des cas sont asymptomatiques. Néanmoins, dans une étude populationnelle regroupant 1 113 sujets âgés en moyenne de 88,5 ans, 78,9 % avaient les critères anatomopathologiques d'AAC à l'autopsie mais les deux tiers d'entre eux avaient une charge lésionnelle considérée comme seulement légère[1]. La prévalence de la maladie était la même quel que soit le sexe. Parmi l'ensemble des patients présentant des lésions d'AAC, 41 % avaient également un diagnostic clinique de maladie d'Alzheimer, ce qui prouve la fréquente coexistence de ces deux maladies. Enfin, la fréquence de la maladie, exceptionnelle avant l'âge de 55 ans, augmente avec l'âge, puisque dans une autre étude autoptique[2], la prévalence de lésions sévères d'AAC était de 2,3 % chez les patients de 65 à 74 ans, de 8 % de 75 à 84 ans et de 12 % au delà de 85 ans.
La proportion de patients présentant une AAC pathologique qui développent une AAC clinique (c'est-à-dire qui présenteront une hémorragie cérébrale) n'est pas connue. Néanmoins, dans le registre des hémorragies cérébrales de Helsinki, 20 % des cas étaient attribués à l'AAC ce qui en faisait la seconde cause d'hémorragie cérébrale de l'adulte après la microangiopathie hypertensive qui représenterait 35 % des cas[3]. Il semble également que la proportion des hémorragies cérébrales liées à l'AAC soit en forte augmentation au cours de ces trente dernières années ce qui s'expliquerait par trois facteurs : le meilleur contrôle de l'hypertension artérielle (réduisant la part des hémorragies liées à la microangiopathie hypertensive), le vieillissement de la population et le recours de plus en plus fréquent aux fluidifiants du sang (anticoagulants et antiagrégants plaquettaires) qui majorent le risque hémorragique[4].
Physiopathologie
Histologie
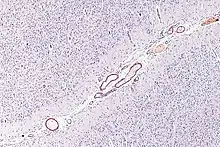
L'AAC résulte du dépôt d'amylose dans la paroi des vaisseaux cérébraux mis en évidence par une coloration au Rouge Congo. La forme d'AAC a plus répandue est l'AAC sporadique β-amyloïde qui résulte du dépôt de peptide Aβ-40 dans la média et l'adventice des artérioles des vaisseaux corticaux ou leptoméningés. Néanmoins, d'autres formes moléculaires ont été décrites (par exemple liées au dépôt de Cystatine C ou de Transthyrétine) dont la fréquence exacte est inconnue en raison de leur grande rareté[5].
Le dépôt du peptide Aβ-40 débute généralement dans la membrane basale externe puis s'étend vers la média des vaisseaux, ce qui cause la dégénération des cellules musculaires lisses qui s'y trouvent. Dans les formes sévères, la média et l'adventice ont disparu et sont remplacées par de la nécrose amyloïde. Seul l'endothelium, paroi la plus fine et la plus interne du vaisseau, reste intacte. Cette forme sévère est souvent associée avec des dilatations microanévrysmales ou des occlusions du vaisseau. On distingue généralement deux formes de AAC selon que les capillaires sont également atteints ou non. En règle générale, l'atteinte prédomine sur la partie postérieure du cerveau et les vaisseaux profonds du cerveau sont respectés[6].
Pathogenèse
L'AAC partage une voie physiopathologique avec la maladie d'Alzheimer dans la mesure où le peptide Aβ-40 de l'AAC et le peptide Aβ-42 de la maladie d'Alzheimer sont l'un et l'autre issus du clivage de la protéine APP (pour Amyloid percursor protein) par une famille d'enzymes, les « β-sécretase » et « γ-sécretases », La différence entre les deux peptides résulte d'une différence dans le nombre d'acide-aminé : 40 dans le cas de l'Aβ-40 et 42 dans le cas de Aβ-42. De ce fait, l'Aβ-40 est plus soluble que Aβ-42 et est transportée — à travers les réseaux de drainage du tissu interstitiel — vers les vaisseaux sanguins où ils se déposent. Ils sont alors responsables : soit d'une fragilisation de la paroi du vaisseau qui peut se rompre causant une hémorragie cérébrale, soit de l'occlusion de petits vaisseaux qui causent de microinfarctus cérébraux et par là-même d'une perte neuronale (pouvant expliquer les troubles cognitifs observés dans la maladie).
Manifestations cliniques
- Hémorragie cérébrale : la principale manifestation clinique de l'angiopathie amyloïde est l'hémorragie intracérébrale de topographie lobaire ou cérébelleuse[7]. La topographie lobaire regroupe les structures corticales et sous-corticale (c'est-à-dire proche de la surface du cerveau). Elle est principalement définie par opposition à la topographie profonde qui englobe essentiellement les ganglions de la base (en particulier le thalamus ou le noyau lenticulaire) et le pont. Cette distinction topographique est importante dans la mesure où l'hémorragie cérébrale lobaire est très évocatrice d'AAC alors que l'hémorragie cérébrale profonde est plutôt évocatrice de microangiopathie hypertensive. Cette distribution reflète l'atteinte préférentielle des petits vaisseaux corticaux dans l'AAC et de artères lenticulo-striées dans l'angiopathie hypertensive[8]. Cependant, parmi l'ensemble des topographies lobaires possibles, l'atteinte du lobe temporal ou occipital semble prédominante, en particulier si le patient avait déjà présenté une hémorragie dans ces régions[9]. Comme pour l'ensemble des AVC, les symptômes associés aux hémorragies cérébrales de l'AAC sont variables. Ils dépendent avant tout de la taille de l'hémorragie et de sa localisation.
 Scanner cérébral objectivant une hémorragie lobaire temporale droite évocatrice d'une possible angiopathie amyloïde
Scanner cérébral objectivant une hémorragie lobaire temporale droite évocatrice d'une possible angiopathie amyloïde Scanner cérébral objectivant une hémorragie profonde (thalamique gauche) plutôt évocatrice d'une angiopathie hypertensive
Scanner cérébral objectivant une hémorragie profonde (thalamique gauche) plutôt évocatrice d'une angiopathie hypertensive
- Manifestations neurologiques transitoires : l'AAC peut s'accompagner de troubles neurologiques - le plus souvent sensitifs ou moteurs - qui dans leur forme typique peuvent récidiver durant plusieurs semaines (« amyloid spells » dans la littérature scientifique anglo-saxonne). Le mécanisme exact de ces troubles reste inconnu et peuvent être aisément confondus avec des crises d'épilepsie focale ou des accidents ischémiques transitoires. À l'imagerie, ils sont souvent associés à la présence d'hémosidérose voire d'hémorragie sous-arachnoïdienne focale, ne touchant qu'un seul sillon cortical[10].
- Troubles cognitifs : la présence de troubles cognitifs spécifiques de l'AAC n'a été soulignée que récemment. Ceci explique un double facteur de confusion qui a pu masquer les troubles cognitifs propres à l'AAC : d'une part, la fréquente association clinique avec la maladie d'Alzheimer, d'autre part, le fait que l'AAC n'a souvent été étudiée que dans le cadre de l'hémorragie cérébrale, souvent responsable de troubles cognitifs potentiellement sévères par elle-même[11]. Néanmoins, il a été montré dans l'étude autoptique déjà mentionnée plus haut que la présence de lésions histologiques d'AAC est associée à un sur-risque de déclin cognitif indépendamment des autres lésions, en particulier celles de maladie Alzheimer[1]. Les études in vivo rendent plus difficile le contrôle de la part des troubles attribuables à la maladie d'Alzheimer. Une étude chez des patients ayant présenté une hémorragie cérébrale montrait un taux plus élevé de démence à un an chez les patients avec une hémorragie lobaire et de l'hémosidérose corticale et des microsaignements (trois signes clés à l'IRM du diagnostic d'AAC)[12]. Le profil cognitif propre à l'AAC est difficile à déterminer. Comparés à des patients souffrant de maladie d'Alzheimer, les patients avec une AAC (sans antécédent hémorragique) avaient des performances significativement meilleures sur les scores de mémoire épisodique mais comparables pour ceux des fonctions exécutives et de vitesse de traitement[13].
Diagnostic
Critères de Boston
Le diagnostic de l'AAC se fonde sur les critères de Boston de 1995[14] modifiés en 2010[15]. Ces critères permettent de classer les patients en fonction du degré de probabilité du diagnostic. Le diagnostic de certitude est uniquement autoptique. Il est néanmoins possible de poser le diagnostic d'AAC possible ou probable du vivant du patient en s'appuyant sur un faisceau d'arguments essentiellement fondé sur les marqueurs hémorragiques à l'IRM cérébrale qu'il s'agisse de lésion en faveur d'une macrohémorragie cérébrale (> 1 cm de diamètre) ou de microhémorragie (diamètre < 1 cm).
| Niveau de certitude | Critères de Boston modifiés (2010) |
|---|---|
| AAC certaine |
|
| AAC probable avec preuve anatomopathologique |
|
| AAC probable | Données clinique et/ou d'imagerie :
|
| AAC possible | Données clinique et/ou d'imagerie :
|
Validité des critères
La validité des critères de Boston a été validée dans plusieurs séries anatomopathologiques avec une spécificité excellente à chaque fois supérieure à 95 % pour les cohortes hospitalières chez des patients ayant présenté une macrohémorragie cérébrale[16]. En revanche, dans des populations sans évènement macrohémorragique (donc présentant seulement deux microhémorragies lobaires au minimum), la spécificité chutait à moins de 90 %[17]. Par ailleurs, la sensibilité des critères de Boston reste médiocre à 70 % même avec la modification des critères de 2010 qui améliorent la sensibilité (de 57 % à 71 %) sans pour autant en réduire la spécificité[15].
Autres marqueurs diagnostiques d'intérêt
Afin d'augmenter la sensibilité des critères de Boston, plusieurs marqueurs d'intérêts ont été évalués: des marqueurs d'imagerie en IRM et en TEP (tomographie par émission de positons) mais également biologiques à travers les marqueurs de protéines amyloïdes dans le liquide cérébrospinal (LCS).
- Marqueurs en IRM cérébrale. Outre les marqueurs hémorragiques déjà mentionnés, la valeur diagnostique de quasi tous les marqueurs classiques de microangiopathie à l'IRM ont été étudiés, en particulier pour différencier l'AAC de l'angiopathie hypertensive. En particulier, plusieurs travaux se sont intéressés à la distribution des hypersignaux de la substance blanche. L'AAC semble se caractériser par une tendance à présenter des hypersignaux de substance blanche plutôt cortico-sous-corticale (proche du cortex) alors que l'angiopathie hypertensive touche plus spécifiquement les capsules externes et le tronc cérébral[18]. La présence d'hypersignaux punctiformes en IRM de diffusion a également été considéré comme un marqueur potentiel de l'AAC, mais cette association n'a pas été retrouvée dans une méta-analyse de 2018[19]. À l'heure actuelle, seule la présence de dilatation des espaces de Virchow-Robin de topographie sous-corticale a montré une bonne corrélation avec les données histologiques.
- Marqueurs biologiques. Seuls les marqueurs dans le LCR obtenu à partir de ponctions lombaires ont été analysés, et plus particulièrement les marqueurs validés dans la maladie d'Alzheimer, à savoir les marqueurs de la protéine amyloïde (Aβ-40 et Aβ-42) et les marqueurs de protéine tau. Dans une méta-analyse de 2018, le profil de ces marqueurs chez les patients avec une AAC était similaire à celui des patients avec une maladie d'Alzheimer, à l'exception de la protéine Aβ-40, très abaissée dans l'AAC. Néanmoins cette étude n'incluait que 59 patients et aucune corrélation avec l'histologie n'était faite[20].
- Tomographie par émission de positons. Là encore, ce sont les radioligands de la protéine amyloïde qui ont été étudiés avec une valeur diagnostique convenable mais variable selon les traceurs utilisés. La corrélation avec les données histologiques ne sont pas connues. De plus, le seul ligand ayant reçu une autorisation de mise sur le marché (le Florbetapir) n'a pas obtenu un avis favorable de la commission de transparence de la Haute Autorité de Santé[21]. Il n'est donc pas pris en charge pas la sécurité sociale. Pour cette raison, en France comme dans la majorité des pays européens, l'utilisation des techniques de TEP-amyloïde est réservée quasi exclusivement à la recherche.
Pronostic et traitements
il n'y a pas de prise en charge spécifique à l'AAC en cas d’hémorragie. La prise en charge des AAC est celle des AVC hémorragiques en général et repose sur la baisse précoce et rapide de la tension artérielle dès que possible afin de limiter le risque d'extension du volume de l'hématome dans les premières heures ce qui est une complication fréquente des hémorragies cérébrales. Ce traitement repose essentiellement sur l'administration intra-veineuse de médicaments antihypertenseurs (en particulier la Nicardipine) avec un objectif tensionnel inférieur à 140/90 mmHg[22]. Selon la gravité de l'hématome et sa localisation, il convient aussi de lutter contre les complications de décubitus (thromboses veineuse profondes, escarres, dénutrition), et de prendre en charge les troubles de la déglutition. Enfin, si le patient reçoit un traitement anticoagulant, une antagonisation est nécessaire[23]. Les principaux facteurs de mauvais pronostic de l'hémorragie cérébrale sont l'âge (supérieur à 80ans), le score de vigilance défini par l'échelle de Glasgow (un score initial <13), le volume de l'hémorragie (>30ml), la présence initiale d'un drainage de l'hémorragie dans les ventricules cérébraux et une atteinte infratentorielle (touchant le tronc cérébral ou cervelet)[24].
La principale spécificité de la prise en charge de l'AAC dans la prise en charge du risque hémorragique. Les principaux facteurs prédictifs du risque hémorragique semblent être l'antécédent d'hémorragie cérébrale et la présence d'hémosidérose corticale. Dans une étude incluant plus de 300 patients avec une AAC sporadique, le risque de récidive était de 8,9% par an[25]. Il semble exister un sur-risque chez les patients avec au moins un allèle ε2 ou ε4 du gène de l'Apolipoprotéine E[26]. Le risque d'hémorragie en cas d'hémosidérose corticale était de 25 % à 4 ans dans une étude récente incluant 118 patients avec une AAC[27]. En raison de cet important risque hémorragique, la question de l''introduction ou de la poursuite des traitements fluidifiants du sang (anticoagulant et antiagrégants plaquettaires) chez les patients avec une AAC probable se pose. Il semble y avoir un consensus actuellement sur la contre-indication des traitements anticoagulants chez les patients avec une AAC à haut risque et une arythmie cardiaque[28] d'autant plus si les patients sont éligibles à une exclusion chirurgicale de l'auricule (traitement du risque ischémique alternatif à l'anticoagulation dans la fibrillation auriculaire)[29]. Il n'y a pas de consensus à l'heure actuelle sur l'anticoagulation chez les patients ayant un AAC se manifestant uniquement par des microsaignements, même si le risque hémorragique augmente avec leur nombre chez les patients anticoagulés[30]. En l'absence de traitement spécifique de l'AAC, la prévention du risque hémorragique passe avant tout par le strict contrôle tensionnel au long cours, qui a montré un effet favorable sur la récidive hémorragique, même chez les patients avec une hémorragie lobaire[31].
Notes et références
- (en) Patricia A. Boyle, Lei Yu, Sukriti Nag et Sue Leurgans, « Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons », Neurology, vol. 85, no 22, , p. 1930–1936 (ISSN 0028-3878 et 1526-632X, PMID 26537052, PMCID PMC4664125, DOI 10.1212/WNL.0000000000002175, lire en ligne, consulté le )
- (en) S. M. Greenberg et J. P. Vonsattel, « Diagnosis of cerebral amyloid angiopathy. Sensitivity and specificity of cortical biopsy », Stroke, vol. 28, no 7, , p. 1418-1422 (ISSN 0039-2499, PMID 9227694, lire en ligne, consulté le )
- (en) Meretoja Atte, Strbian Daniel, Putaala Jukka et Curtze Sami, « Smash-u », Stroke, vol. 43, no 10, , p. 2592-2597 (DOI 10.1161/STROKEAHA.112.661603, lire en ligne, consulté le )
- (en) Maurice Giroud, Olivier Rouaud, Corine Aboa-Eboulé et Jérôme Durier, « Intracerebral haemorrhage profiles are changing: results from the Dijon population-based study », Brain, vol. 136, no 2, , p. 658–664 (ISSN 0006-8950, DOI 10.1093/brain/aws349, lire en ligne, consulté le )
- (en) Masahito Yamada et Hironobu Naiki, « Cerebral Amyloid Angiopathy », dans Progress in Molecular Biology and Translational Science, vol. 107, Elsevier, (ISBN 9780123858832, DOI 10.1016/b978-0-12-385883-2.00006-0., lire en ligne), p. 41–78
- (en) Masahito Yamada, « Cerebral Amyloid Angiopathy: Emerging Concepts », Journal of Stroke, vol. 17, no 1, , p. 17–30 (ISSN 2287-6391 et 2287-6405, PMID 25692104, PMCID PMC4325636, DOI 10.5853/jos.2015.17.1.17, lire en ligne, consulté le )
- (en) Y. Itoh, M. Yamada, M. Hayakawa et E. Otomo, « Cerebral amyloid angiopathy: a significant cause of cerebellar as well as lobar cerebral hemorrhage in the elderly », Journal of the Neurological Sciences, vol. 116, no 2, , p. 135-141 (ISSN 0022-510X, PMID 8336159, lire en ligne, consulté le )
- (en) Andreas Charidimou, Gregoire Boulouis, Kellen Haley et Eitan Auriel, « White matter hyperintensity patterns in cerebral amyloid angiopathy and hypertensive arteriopathy », Neurology, vol. 86, no 6, , p. 505-511 (ISSN 1526-632X, PMID 26747886, PMCID PMCPMC4753727, DOI 10.1212/WNL.0000000000002362, lire en ligne, consulté le )
- (en) Jonathan Rosand, Alona Muzikansky, Ashok Kumar et Jonathan J. Wisco, « Spatial clustering of hemorrhages in probable cerebral amyloid angiopathy », Annals of Neurology, vol. 58, no 3, , p. 459-462 (ISSN 0364-5134, PMID 16130107, DOI 10.1002/ana.20596, lire en ligne, consulté le )
- (en) Charidimou Andreas, Peeters Andre, Fox Zoe et Gregoire Simone M., « Spectrum of Transient Focal Neurological Episodes in Cerebral Amyloid Angiopathy », Stroke, vol. 43, no 9, , p. 2324-2330 (DOI 10.1161/STROKEAHA.112.657759, lire en ligne, consulté le )
- (en) M. Planton, N. Raposo, J.-F. Albucher et J. Pariente, « Cerebral amyloid angiopathy-related cognitive impairment: The search for a specific neuropsychological pattern », Revue Neurologique, vol. 173, no 9, , p. 562–565 (DOI 10.1016/j.neurol.2017.09.006, lire en ligne, consulté le )
- (en) Charlotte Cordonnier, Didier Leys, Alain Duhamel et Hilde Hénon, « Dementia risk after spontaneous intracerebral haemorrhage: a prospective cohort study », The Lancet Neurology, vol. 15, no 8, , p. 820–829 (ISSN 1474-4422 et 1474-4465, PMID 27133238, DOI 10.1016/S1474-4422(16)00130-7, lire en ligne, consulté le )
- (en) Nevicia F. Case, Anna Charlton, Angela Zwiers et Saima Batool, « Cerebral Amyloid Angiopathy Is Associated With Executive Dysfunction and Mild Cognitive Impairment », Stroke, vol. 47, no 8, , p. 2010-2016 (ISSN 0039-2499 et 1524-4628, DOI 10.1161/strokeaha.116.012999, lire en ligne, consulté le )
- (en) Steven M. Greenberg, G. William Rebeck, Jean Paul G. Vonsattel et Teresa Gomez-Isla, « Apolipoprotein E ϵ4 and cerebral hemorrhage associated with amyloid angiopathy », Annals of Neurology, vol. 38, no 2, , p. 254–259 (DOI 10.1002/ana.410380219, lire en ligne, consulté le )
- (en) J. Linn, A. Halpin, P. Demaerel et J. Ruhland, « Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy », Neurology, vol. 74, no 17, , p. 1346–1350 (ISSN 0028-3878 et 1526-632X, PMID 20421578, PMCID PMC2875936, DOI 10.1212/WNL.0b013e3181dad605, lire en ligne, consulté le )
- (en) Steven M. Greenberg et Andreas Charidimou, « Diagnosis of Cerebral Amyloid Angiopathy: Evolution of the Boston Criteria », Stroke, vol. 49, no 2, , p. 491–497 (ISSN 0039-2499 et 1524-4628, DOI 10.1161/STROKEAHA.117.016990, lire en ligne, consulté le )
- (en) Sergi Martinez-Ramirez, Jose-Rafael Romero, Ashkan Shoamanesh et Ann C. McKee, « Diagnostic value of lobar microbleeds in individuals without intracerebral hemorrhage », Alzheimer's & Dementia, vol. 11, no 12, , p. 1480–1488 (PMID 26079413, PMCID PMC4677060, DOI 10.1016/j.jalz.2015.04.009, lire en ligne, consulté le )
- (en) Andreas Charidimou, Gregoire Boulouis, Kellen Haley et Eitan Auriel, « White matter hyperintensity patterns in cerebral amyloid angiopathy and hypertensive arteriopathy », Neurology, vol. 86, no 6, , p. 505-511 (ISSN 0028-3878, PMID 26747886, PMCID PMCPMC4753727, DOI 10.1212/WNL.0000000000002362, lire en ligne, consulté le )
- (en) Boulanger Marion, Schneckenburger Romain, Join-Lambert Claire et Werring David J., « Diffusion-Weighted Imaging Hyperintensities in Subtypes of Acute Intracerebral Hemorrhage », Stroke, vol. 50, no 1, , p. 135-142 (DOI 10.1161/STROKEAHA.118.021407, lire en ligne, consulté le )
- (en) Andreas Charidimou, Jan O. Friedrich, Steven M. Greenberg et Anand Viswanathan, « Core cerebrospinal fluid biomarker profile in cerebral amyloid angiopathy: A meta-analysis », Neurology, vol. 90, no 9, , e754-e762 (ISSN 1526-632X, PMID 29386280, PMCID PMCPMC5837868, DOI 10.1212/WNL.0000000000005030, lire en ligne, consulté le )
- « Haute Autorité de Santé - AMYVID (florbétapir), produit diagnostique pour le système nerveux central », sur has-sante.fr (consulté le )
- (en) Craig S. Anderson, Emma Heeley, Yining Huang et Jiguang Wang, « Rapid Blood-Pressure Lowering in Patients with Acute Intracerebral Hemorrhage », New England Journal of Medicine, vol. 368, no 25, , p. 2355–2365 (ISSN 0028-4793 et 1533-4406, DOI 10.1056/NEJMoa1214609, lire en ligne, consulté le )
- Hemphill J. Claude, Greenberg Steven M., Anderson Craig S. et Becker Kyra, « Guidelines for the Management of Spontaneous Intracerebral Hemorrhage », Stroke, vol. 46, no 7, , p. 2032–2060 (DOI 10.1161/STR.0000000000000069, lire en ligne, consulté le )
- (en) Solène Moulin et Charlotte Cordonnier, « Prognosis and Outcome of Intracerebral Haemorrhage », dans Frontiers of Neurology and Neuroscience, vol. 37, S. Karger AG, (ISBN 9783318055962, DOI 10.1159/000437122, lire en ligne), p. 182-192
- (en) Ellis S. van Etten, M. Edip Gurol, Jeroen van der Grond et Joost Haan, « Recurrent hemorrhage risk and mortality in hereditary and sporadic cerebral amyloid angiopathy », Neurology, vol. 87, no 14, , p. 1482–1487 (ISSN 0028-3878 et 1526-632X, PMID 27590282, PMCID PMC5075977, DOI 10.1212/WNL.0000000000003181, lire en ligne, consulté le )
- (en) Heather C. O'Donnell, Jonathan Rosand, Katherine A. Knudsen et Karen L. Furie, « Apolipoprotein E Genotype and the Risk of Recurrent Lobar Intracerebral Hemorrhage », New England Journal of Medicine, vol. 342, no 4, , p. 240–245 (ISSN 0028-4793 et 1533-4406, DOI 10.1056/NEJM200001273420403, lire en ligne, consulté le )
- (en) David John Werring, Jean-Claude Baron, Patrice Laloux et Yves Vandermeeren, « Cortical superficial siderosis and intracerebral hemorrhage risk in cerebral amyloid angiopathy », Neurology, vol. 81, no 19, , p. 1666–1673 (ISSN 0028-3878 et 1526-632X, PMID 24107862, DOI 10.1212/01.wnl.0000435298.80023.7a, lire en ligne, consulté le )
- (en) Rocco J. Cannistraro et James F. Meschia, « The Clinical Dilemma of Anticoagulation Use in Patients with Cerebral Amyloid Angiopathy and Atrial Fibrillation », Current Cardiology Reports, vol. 20, no 11, , p. 106 (ISSN 1534-3170, DOI 10.1007/s11886-018-1052-1, lire en ligne, consulté le )
- (en) Ricardo Kosturakis et Matthew J. Price, « Current State of Left Atrial Appendage Closure », Current Cardiology Reports, vol. 20, no 6, , p. 42 (ISSN 1534-3170, PMID 29680999, DOI 10.1007/s11886-018-0981-z, lire en ligne, consulté le )
- (en) Emma Young, Inez Wynter, Belinda Wroath et Sarah Wilson-Owen, « Cerebral microbleeds and intracranial haemorrhage risk in patients anticoagulated for atrial fibrillation after acute ischaemic stroke or transient ischaemic attack (CROMIS-2): a multicentre observational cohort study », The Lancet Neurology, vol. 17, no 6, , p. 539–547 (ISSN 1474-4422 et 1474-4465, PMID 29778365, DOI 10.1016/S1474-4422(18)30145-5, lire en ligne, consulté le )
- (en) PROGRESS Collaborative Group, « Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack », Lancet, vol. 358, no 9287, , p. 1033-1041 (ISSN 0140-6736, PMID 11589932, DOI 10.1016/S0140-6736(01)06178-5, lire en ligne, consulté le )
 Portail de la médecine
Portail de la médecine