Carcinome à cellules de Merkel
Le carcinome à cellules de Merkel est un cancer de la peau. Il est classé parmi les tumeurs neuro-endocrines cutanées rares.
| Médicament | Lambrolizumab et nivolumab |
|---|---|
| Spécialité | Oncologie |
| CIM-10 | C44 |
|---|---|
| CIM-9 | 209.31 |
| ICD-O | 8247/3 et 8190/3 |
| DiseasesDB | 31386 |
| eMedicine | 1100917 |
| MeSH | D015266 |
![]() Mise en garde médicale
Mise en garde médicale
Il a été décrit pour la première fois par Toker en 1972[1].
Fréquence, étiologie
C'est une affection qui touche essentiellement les sujets âgés (95 % des cas après 50 ans) à peau claire avec une répartition équivalente dans les deux sexes ; sa fréquence est inférieure à 1/200 000 mais son incidence a été multiplié par 4, aux États-Unis, en 1986 et 2011[2]. Il est le plus souvent observé dans les zones du revêtement cutané les plus photo-exposées[3] ; un autre facteur de risque est le déficit immunitaire (immuno-suppression chez les greffés d'organe ou secondaire à une infection par VIH)[4].
En 2008, a été mis en évidence l'existence de séquences virales au sein des cellules de ce cancer[5] (Merkel cell polyomavirus (en)) ; d'autres chercheurs ont confirmé cette découverte[6]. Ce polyomavirus exprime des antigènes qui peuvent être recherchés et constituent un marqueur de l'extension de la maladie[7].
Les formes sans polyomavirus sont essentiellement dues à l'exposition solaire et les cellules concernées comportent alors de nombreuses mutations qui ne sont pas retrouvées dans les formes avec virus[8]. Elles constituent à peu près la moitié des carcinomes à cellules de Merkel[9].
Description clinique, anatomo-pathologie
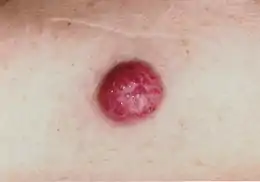
Le carcinome à cellules de Merkel à son origine est une tumeur du derme qui se présente sous la forme d'un nodule dur, isolé, le plus fréquemment aux extrémités, et dans près de la moitié des cas, au niveau de la tête et du cou[10].
Le diagnostic n'est pas aisé : le nodule est souvent mobile, non adhérent aux tissus avoisinants, non douloureux et de forme harmonieuse. Il est discrètement érythémateux, puis deviendra violacé. Sa croissance est rapide, souvent de 1 cm par mois et la tumeur atteindra rapidement les tissus sous-jacents (fascia, muscle)[11]. Le diagnostic n'est réellement fait que par l'anatomo-pathologiste en immunohistochimie. On retrouve des marqueurs neuro-endocrines (énolase neuronale spécifique, synaptophysine et chromogranine A), de la cytokératine 20 et des neurofilaments.
Le Professeur Paul Nghiem de l'université de Washington a créé un acronyme en anglais pour permettre aux médecins de se souvenir des caractéristiques propres à ce carcinome. AEIOU A car c'est une lésion asymptomatique, E car elle s'étend rapidement, I pour immunodéprimé, O pour older than 50 years (plus âgé que 50 ans) et finalement U pour l'exposition aux UV et les peaux claires[12].
Pronostic
C'est un cancer lymphophile.
Le pronostic de ce cancer est assez sombre : il est actuellement considéré comme plus réservé que dans le cas du mélanome. On distingue 4 stades dans l'évolution de la maladie (classification AJCC) :
- stade I : lésion primitive inférieure à 2 cm,
- stade II : lésion primitive supérieure à 2 cm,
- stade III: présence d'un ganglion métastatique,
- stade IV : présence de métastase à distance.
Les malades indemnes de lésion ganglionnaire ont un taux de survie à cinq ans supérieur à 80 %, mais ce taux est inférieur à 50 % en cas d'atteinte ganglionnaire. La présence de métastases ganglionnaires, pouvant être mises en évidence par la technique du repérage du ganglion sentinelle[13], assombrit ce pronostic : le risque de rechute est de 60 % dans les trois ans en cas de présence de micrométastases lymphatiques, alors qu'il n'est que de 20 % en l'absence de micro-métastase. De rares cas de régression spontanée par apoptose des cellules tumorales ont été décrits[14].
Si le carcinome est une forme à polyomavirus, la négativation sous traitement des anticorps contre une protéine de ce virus (oncoprotéine MCPyV) pourrait être un indice de bon pronostic[9].
Traitement
La prise en charge thérapeutique dépend du stade de l'évolution du cancer au moment du diagnostic. Il convient donc de pratiquer un bilan de l'extension de la maladie. Ce bilan, outre un examen clinique complet comportera une échographie des aires ganglionnaires, une scanner thoraco-abdomino-pelvien et un scanner cérébral. Selon les cas, on pourra discuter une scintigraphie osseuse, voire un petscan.
- Aux stades I et II, on pratique une exérèse chirurgicale de la lésion en passant très "au large", soit avec des marges de 3 cm (supérieures aux marges pratiquées en cas de mélanome). La radiothérapie locale diminue l'incidence des récidives locales[15], et semble même aussi efficace seule qu'associée à la chirurgie[16]. On y associe classiquement une radiothérapie des gites ganglionnaires en cas de micro-métastase à l'examen du ganglion sentinelle, mais les preuves en termes d'amélioration de la survie font défaut.
- Au stade III avec atteinte ganglionnaire avérée, on pratique l'exérèse de la lésion et on y associe un curage ganglionnaire. Cette chirurgie est complétée par une radiothérapie du site ganglionnaire. S'il s'agit d'un récidive, la chimiothérapie se discute, selon l'état général du patient. Le pembrolizumab, un inhibiteur du PD1 semble donner des résultats prometteurs[17] et l'avélumab serait efficace dans les formes réfractaires[18].
- Au stade IV, on peut retrouver des métastases au niveau du foie, des os, du poumon, du cerveau ou de la peau. Le traitement est alors palliatif associant radio et chimiothérapie. Une exérèse chirurgicale d'une métastase unique est envisageable.
Perspectives d'avenir
Compte tenu du vieillissement de la population, son incidence devrait augmenter et afin d'optimiser sa prise en charge, des centres de référence ont été créés[19].
Notes et références
- Toker C. Trabecular carcinoma of the skin Arch. Dermatol. 1972 ; 105 : 109-110
- Fitzgerald TL, Dennis S, Kachare SD, Vohra NA, Wong JH, Zervos EE, Dramatic increase in the incidence and mortality from Merkel cell carcinoma in the United States, Am Surg, 2015;81:802-806
- Heath M., Jaimes N., Lemos B., Mostaghimi A., Wang L.C., Peñas P.F., et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features J Am Acad Dermatol. 2008 ; 58 : 375-381
- Engels E.A., Frisch M., Goedert J.J., Biggar R.J., Miller R.W. Merkel cell carcinoma and HIV infection Lancet. 2002 ; 359 : 497-498
- Feng H, Shuda M, Chang Y, Moore PS, Clonal integration of a polyomavirus in human Merkel cell carcinoma, Science, 2008;319:1096-1100
- Découverte du MPCPyV dans un cancer à cellules de Merkel en France
- Paulson KG, Carter JJ, Johnson LG et al. Antibodies to Merkel cell polyomavirus T antigen oncoproteins reflect tumor burden in Merkel cell carcinoma patients, Cancer Res, 2010;70:8388-8397
- Harms PW, Vats P, Verhaegen ME et al. The distinctive mutational spectra of polyomavirus-negative Merkel cell carcinoma, Cancer Res, 2015;75:3720-3727
- Paulson KG, Lewis CW, Redman MW et al. Viral oncoprotein antibodies as a marker for recurrence of Merkel cell carcinoma: a prospective validation study, Cancer, 2017;123:1464-1474
- Grabowski J, Saltzstein SL, Sadler GR, Tahir Z, Blair S, A comparison of Merkel cell carcinoma and melanoma: results from the California cancer registry, Clin Med Oncol, 2008;2:327-333
- Description clinique, voir Histoire naturelle
- Heath, M., Jaimes, N., Lemos, B., Mostaghimi, A., Wang, L. C., Peñas, P. F., & Nghiem, P. (2008). Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. Journal of the American Academy of Dermatology, 58(3), 375-381.
- Description de la technique dans le cas du cancer du sein
- Description d'un cas de régression spontanée
- Amélioration du taux de survie après radiothérapie locale
- Résultats de la radiothérapie isolée chez des patients inopérables
- Nghiem PT, Bhatia S, Lipson EJ et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma, N Engl J Med, 2016;374:2542-2552
- Kaufman HL, Russell J, Hamid O et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma, Lancet Oncol, 2016;17:1374-1385
- Centres de référence en France
 Portail de la médecine
Portail de la médecine