Chaîne idéale
La chaîne idéale est le modèle le plus simple utilisé pour décrire les polymères[1], comme les acides nucléiques et les protéines. Le polymère est considéré comme une marche aléatoire[2], et toute interaction entre les monomères est négligée. Bien que ce modèle soit élémentaire, il donne un aperçu de la physique des polymères.
Dans ce modèle, les monomères sont des tiges rigides de longueur fixée l, et leur orientation est complètement indépendante des positions et orientations de leurs voisins, dans la mesure où deux polymères peuvent coexister à la même place. Dans certains cas, le monomère peut être interprété physiquement comme un amino-acide dans un polypeptide. Dans d'autres cas, un monomère est simplement un segment de polymère, représenté comme une unité libre, discrète et donc, l est la longueur de Kuhn. Par exemple, la chromatine est modélisée comme un polymère, pour lequel chaque segment mesure environ 14 à 46 kilobases de long[3].
Modèle
Soit un polymère constitué de N unités monomères, dont la longueur totale une fois déplié est :
- .

Dans cette approche simple, où l'on considère que les monomères ne sont pas en interaction, l'énergie du polymère est choisie de telle sorte qu'elle soit indépendante de sa forme. Ceci qui signifie qu'à l'équilibre thermodynamique, toutes les configurations du polymère ont autant de chances les unes que les autres de s'établir lors du mouvement du polymère, relativement à la distribution de Maxwell–Boltzmann.
Soit le vecteur d'un bout à l'autre de la chaîne idéale, et les vecteurs correspondant à chaque monomère. Ces vecteurs aléatoires ont des composantes dans les trois directions de l'espace. La plupart des expressions de cet article suivent l'hypothèse que le nombre de monomères est grand, c'est pourquoi le théorème central limite s'applique. La figure ci-dessous représente le schéma d'une chaîne idéale (courte).



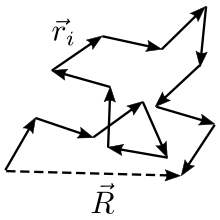
Les deux extrémités de la chaîne ne coïncident pas, mais fluctuent l'une autour de l'autre, d'où :
- .

Tout au long de cet article, les symboles désigneront la valeur moyenne (par rapport au temps) de variables ou vecteurs aléatoires, comme ci-dessus.

Comme sont indépendants, il vient du théorème central limite que suit une distribution normale[4] (ou distribution de Gauss) : précisément, en 3D, et suivent une distribution normale de moyenne et de variance :



- .
- .


Et ainsi : . Le vecteur d'un bout à l'autre de la chaîne suit la fonction de densité de probabilité suivante :

- .

La distance moyenne d'une extrémité à l'autre du polymère est donnée par :
- .

Le rayon de giration est une quantité fréquemment utilisée en physique des polymères, et peut s'exprimer comme suit :
- .

Il est important de noter que la distance moyenne d'une extrémité à l'autre du polymère, donnée ci-dessus, qui dans le cas de notre modèle simplifié représente typiquement l'amplitude des fluctuations du système, devient négligeable comparée à la longueur totale du polymère déplié à la limite thermodynamique. Ce résultat est une propriété générale des systèmes statistiques.
Remarque mathématique : la démonstration rigoureuse de l'expression de la densité de probabilité n'est pas si évidente qu'elle l'est apparue précédemment : de l'application du théorème central limite, il apparaît que et suivent une loi de distribution normale centrée de la variance . L'expression donnée ci-dessus pour n'est pas la seule qui soit compatible avec une telle distribution pour et . Cependant, comme les composantes des vecteurs sont décorrélées pour le trajet aléatoire que nous considérons, il vient que et sont également décorrélés. Cette condition additionnelle peut uniquement être remplie si suit la distribution de . Autrement, ce résultat peut être démontré par l'application d'une généralisation multidimensionnelle du théorème central limite, ou par des arguments de symétrie.


Généralités du modèle
Alors que le modèle élémentaire décrit ci-dessus est totalement inapproprié pour la description des polymères réels à l'échelle microscopique, il peut cependant faire preuve de pertinence à l'échelle macroscopique dans le cas d'un polymère en solution, dont les monomères forment un mélange idéal avec le solvant (car dans ce cas, les interactions entre monomère et monomère, entre molécule de solvant et molécule de solvant, et entre monomère et molécule de solvant sont identiques, de telle sorte que l'énergie du système peut être considérée constante, validant les hypothèses du modèle). La pertinence de ce modèle est, malgré cela, limitée à l'échelle macroscopique, par le fait qu'il néglige les effets stériques (c'est-à-dire qu'il ne prend pas en compte la place qu'occupe chaque atome dans une molécule).
Les autres modèles de polymères fluctuants ne prenant pas en compte les interactions entre monomères ni les effets stériques, comme le worm like chain model, convergent tous asymptotiquement vers le modèle de chaîne idéale une fois la limite thermodynamique atteinte. Pour cette analogie, on introduit un segment de Kuhn, correspondant à un monomère de longueur équivalente, à considérer dans la chaîne idéale analogue. Le nombre de segments de Kuhn à considérer dans la chaîne idéale analogue est égal au quotient de la longueur totale du polymère déplié, par la longueur d'un segment de Kuhn.
Élasticité entropique d'une chaîne idéale
Si les deux extrémités libres d'une chaîne idéale sont attachées par un dispositif de micro-manipulation, alors le dispositif subit une force exercée par le polymère. L'énergie de la chaîne idéale est constante, et donc sa moyenne temporelle, l'énergie interne, est aussi constante, ce qui signifie que cette force provient nécessairement d'un effet purement entropique.
La force entropique est très similaire à la pression subie par les parois d'une boîte contenant un gaz idéal. L'énergie interne d'un gaz idéal dépend uniquement de sa température, et pas du volume de la boîte qui le contient. Ce n'est donc pas un effet énergétique qui tend à augmenter le volume de la boîte comme le fait la pression du gaz. Ceci implique que la pression d'un gaz idéal a une origine purement entropique[5].
Quelle est donc l'origine microscopique d'une telle force ou d'une telle pression entropiques ? La réponse la plus commune concerne l'effet des fluctuations thermiques, qui tend à amener un système thermodynamique vers un état macroscopique, qui correspond à un maximum dans le nombre d'états microscopiques (ou micro-états) qui sont compatibles avec cet état macroscopique. En d'autres termes, les fluctuations thermiques tendent à amener un système vers son état macroscopique d'entropie maximale.
Que cela signifie-t-il dans le cas de la chaîne idéale ? Tout d'abord, pour la chaîne idéale, un état microscopique est caractérisé par une superposition des états individuels de chaque monomère (avec i variant de 1 à N). Dans son solvant, la chaîne est constamment sujette à des chocs dus aux mouvements des molécules de solvant, et chacun de ces chocs envoie le système de son état microscopique actuel jusqu'à un autre état microscopique, très similaire. Pour un polymère idéal, comme cela a été montré précédemment, il y a plus d'états microscopiques compatibles dans le cas d'une distance d'un bout à l'autre du polymère faible, que dans le cas d'une longue distance. Ainsi, pour une chaîne idéale, maximiser l'entropie signifie réduire la distance entre ses deux extrémités libres. De ce fait, une force qui essaie de plier la chaîne est exercée par la chaîne idéale entre ses deux extrémités[6].

Dans cette section, la moyenne de cette force sera dérivée. L'expression générale obtenue à la limite thermodynamique sera ensuite discutée.
Chaîne idéale sous contrainte de longueur : ensemble de Helmholtz
Dans cette sous-section, nous considérerons le cas d'une chaîne idéale dont les deux extrémités sont attachées à des points fixes. Le vecteur joignant ces deux points caractérise l'état macroscopique (ou macro-état) de la chaîne idéale. Chaque macro-état correspond à un certain nombre de micro-états, que nous appellerons (les micro-états sont définis dans l'introduction de cette section). Comme l'énergie de la chaîne idéale est constante, chacun de ces micro-états a autant de chances qu'un autre de s'établir. L'entropie associée à un macro-état est ainsi égale à :

- , avec la constante de Boltzmann.


L'expression ci-dessus donne l'entropie absolue (quantique) du système. Une détermination précise de requerrait un modèle quantique pour la chaîne idéale, ce qui est au-delà de la portée de cet article. Cependant, nous avons déjà calculé la densité de probabilité associée au vecteur de la totalité de la chaîne non contrainte, ci-dessus. Comme tous les micro-états de la chaîne idéale ont la même probabilité de s'établir, est proportionnel à . Ceci nous conduit à l'équation suivante, pour l'expression de l'entropie classique (relative) de la chaîne idéale :
- , où est une constante fixée.


Soit la force exercée par la chaîne sur le point où son extrémité est attachée. De l'expression précédente de l'entropie, on peut déduire une expression de cette force. Maintenant, supposons qu'au lieu d'être fixée, les positions des deux extrémités de la chaîne idéale soient contrôlées par un opérateur. Les opérateurs contrôlent l'évolution du vecteur entre les extrémités . Si l'opérateur fait varier d'une petite quantité , alors la variation d'énergie interne est nulle, puisque l'énergie de la chaîne est constante. Cette condition peut être écrite comme suit :


- .

est définie comme la quantité élémentaire de travail mécanique transférée par l'opérateur à la chaîne idéale, et est définie comme la quantité élémentaire de chaleur transférée par le solvant à la chaîne idéale. Maintenant, si l'on suppose que la transformation imposée par l'opérateur est quasi-statique (c'est-à-dire infiniment lente), alors la transformation du système sera réversible par rapport au temps, et l'on peut supposer que durant le passage d'un macro-état à un macro-état , le système passe par une série de macro-états à l'équilibre thermodynamique. Ceci a deux conséquences :



- la quantité de chaleur reçue par le système pendant la transformation peut être reliée à la variation de son entropie :
- , où est la température de la chaîne ;


- afin que la transformation reste infiniment lente, la moyenne de la force exercée par l'opérateur sur les extrémités de la chaîne doit équilibrer la moyenne de la force exercée par la force sur ses propres extrémités. Soit la force exercée par l'opérateur et la force exercée par la chaîne, alors on obtient :


- .

Il en vient que :
- .


L'équation ci-dessus est l'équation d'état de la chaîne idéale. Comme l'expression dépend du théorème central limite, il est exact uniquement dans le cas limite d'un polymère contenant un nombre élevé d'unités monomères (donc, à la limite thermodynamique). Il est également seulement valide dans le cas d'une distance entre les extrémités de la chaîne faible par rapport à la longueur totale du contour du polymère, où le comportement est semblable à celui d'un ressort hookéen. Le comportement pour de plus grandes plages de force peut être modélisé en utilisant traitement d'ensemble canonique identique à celui de la magnétisation des spins paramagnétiques. Pour les forces arbitraires, la relation force-extension est donnée par la fonction de Langevin :


où l'extension est .

Pour des extensions arbitraires, la relation force-extension peut être approximée comme suit :
- ,

où est l'inverse de la fonction de Langevin, [7] est le nombre de liaisons dans la molécule (donc si la molécule a liaisons, elle est composée de unités monomères).


Finalement, le modèle peut être étendu pour des plages de force encore plus larges, en incluant un module d'élasticité le long du contour du polymère, c'est-à-dire en permettant à chaque unité monomère de la chaîne de répondre élastiquement à la force appliquée[8].
Chaîne idéale sous contrainte de force : ensemble de Gibbs
À travers cette sous-section, comme dans la précédente, les deux extrémités du polymère sont attachées par un dispositif de micro-manipulation. Cette fois, cependant, le dispositif ne maintient pas les deux extrémités de la chaîne dans une position fixe, mais il les maintient plutôt grâce à une force de traction constante sur la chaîne idéale. Dans ce cas, les positions des deux extrémités de la chaîne fluctuent autour d'une position moyenne . La chaîne idéale réagit par une force constante opposée .


Pour une chaîne idéale dans l'ensemble de Gibbs, sous contrainte de force, un macro-état du système est caractérisé par le vecteur .
La différence entre une chaîne idéale sous contrainte de longueur et une chaîne idéale sous contrainte de force est très proche de la différence entre les ensemble microcanonique et ensemble canonique. Le changement consiste à passer d'un état où une valeur fixe est imposée sur un certain paramètre, à un état où le système est laissé libre d'échanger sur ce paramètre avec l'extérieur. Le paramètre en question est l'énergie, pour les descriptions micro-canonique et canonique, alors qu'il s'agit de la longueur de la chaîne dans le cas d'une chaîne idéale.
Comme dans les ensembles micro-canonique et canonique, les deux descriptions de la chaîne idéale diffèrent seulement par la façon dont sont traitées les fluctuations du système. Ainsi, ils sont équivalents à la limite thermodynamique. L'équation d'état de la chaîne idéale reste la même, excepté qu'elle est désormais sujette à des fluctuations.
Références
- James E. Mark, Physical properties of polymer handbook, Springer, (ISBN 978-0-387-31235-4, 0387312358 et 9780387690025, OCLC 619279219, lire en ligne)
- Meyer B. Jackson, Molecular and cellular biophysics, Cambridge University Press, , 512 p. (ISBN 978-0-521-62441-1, 9780521624701 et 052162441X, OCLC 61757063, lire en ligne)
- Karsten Rippe, « Making contacts on a nucleic acid polymer », Trends in Biochemical Sciences, vol. 26, no 12, , p. 733–740 (ISSN 0968-0004, DOI 10.1016/s0968-0004(01)01978-8, lire en ligne, consulté le )
- Martial Mazars, « Statistical physics of the freely jointed chain », Physical Review E, vol. 53, no 6, , p. 6297–6319 (DOI 10.1103/PhysRevE.53.6297, lire en ligne, consulté le )
- (en) Jerome Harris Weiner, Statistical mechanics of elasticity, New York/Chichester/Brisbane etc., Wiley, , 439 p. (ISBN 0-471-09773-X et 9780471097730, OCLC 8845964, lire en ligne)
- (en) Roland G. Winkler, « Deformation of semi-flexible chains », Journal of Chemical Physics, , p. 2919-2928 (lire en ligne)
- (en) Michael Rubinstein, Polymer physics, Oxford/New York, Oxford University Press, , 440 p. (ISBN 0-19-852059-X, 9780198520597 et 0198520603, OCLC 50339757, lire en ligne)
- S. B. Smith, L. Finzi et C. Bustamante, « Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads », Science, vol. 258, no 5085, , p. 1122–1126 (ISSN 0036-8075, PMID 1439819, lire en ligne, consulté le )
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Ideal chain » (voir la liste des auteurs).