Diffusion Raman
La diffusion Raman, ou effet Raman, est un phénomène optique découvert indépendamment en 1928 par les physiciens Chandrashekhara Venkata Râman et Leonid Mandelstam. Cet effet consiste en la diffusion inélastique d'un photon, c'est-à-dire le phénomène physique par lequel un milieu peut modifier légèrement la fréquence de la lumière qui y circule. Ce décalage en fréquence correspond à un échange d'énergie entre le rayon lumineux et le milieu. Cet effet physique fut prédit par Adolf Smekal en 1923[1].
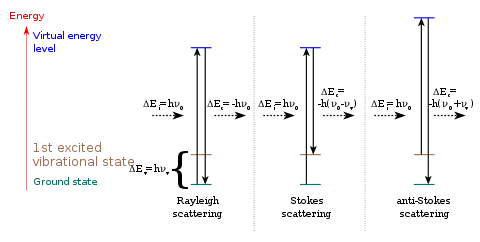
Cet échange peut avoir plusieurs causes : vibrations du cristal ou de la molécule, excitations magnétiques... La mesure de ce décalage permet de remonter à certaines propriétés du milieu. On parle alors de Spectroscopie Raman. Cette technique est largement répandue dans l'industrie et la recherche depuis l'apparition du laser.
Dans le cas particulier où la diffusion est due à des ondes acoustiques, on parle de diffusion Brillouin.
Historique
En 1922, le physicien indien Chandrashekhara Venkata Râman a publié son ouvrage sur « la diffraction moléculaire de la lumière », première d'une série d'investigations avec ses collaborateurs qui a ultimement mené à sa découverte, le 28 février 1928, de l'effet optique qui porte son nom. L'effet Raman a été rapporté pour la première fois par C. V. Raman et K. S. Krishnan (en), et indépendamment par Grigory Landsberg et Leonid Mandelstam en 1928. Raman a reçu le Prix Nobel en 1930 pour son travail sur la diffusion de la lumière. Cette diffusion fut prédite par Adolf Smekal en 1923[1]. Ainsi, dans la littérature allemande, la diffusion de la lumière est-elle nommée effet Smekal-Raman[2].
Description
La diffusion Raman est la diffusion inélastique d'un photon par un milieu. Le fait que la diffusion soit inélastique implique qu'il y a un échange d'énergie entre le photon incident et la molécule via l'excitation vibrationnelle ou bien, en état solide, la création ou l'annihilation d'un phonon optique. Ainsi, la lumière diffusée n'a pas la même longueur d'onde que la lumière incidente. On distingue deux cas :
- décalage Stokes : la lumière est décalée vers le rouge (plus grande longueur d'onde, plus petite énergie) avec l'excitation vibrationnelle ou la création d'un phonon. Le nom Stokes rappelle le physicien George Stokes qui a démontré en 1852 que la fluorescence implique un décalage vers le rouge.
- décalage anti-Stokes : la lumière est décalée vers le bleu (plus courte longueur d'onde, plus grande énergie) avec la désexcitation vibrationnelle ou l'absorption d'un phonon.
S'il n'y a pas d'échange d'énergie entre la molécule et le photon incident, alors la diffusion est élastique et la longueur d'onde du photon diffusé n'est pas décalée. On parle alors de diffusion Rayleigh.
Le décalage en longueur d’onde dépend de la matière et lui est caractéristique : il ne dépend pas de la longueur d’onde d’excitation, ce qui rend possible une analyse de la composition chimique d'un échantillon à partir de la façon dont il diffuse la lumière (voir Application à la spectroscopie Raman ci-dessous).
L'intensité des raies Raman dépend seulement du nombre de molécules dans les différents modes vibrationnels qui leur sont associés. L’utilisation de la distribution de Boltzmann permet de rendre compte correctement du rapport d’intensité entre les raies Stokes et anti-Stokes : les modes vibrationnels de basse énergie étant les plus peuplés, les raies Stokes sont plus intenses que les raies anti-Stokes.
Les photons incidents peuvent être diffusés mais peuvent aussi modifier les vibrations dans l'échantillon étudié, par exemple en état solide par la création (processus Stokes) ou la destruction (processus anti-Stokes) des phonons. Les raies Raman (Stokes ou anti-Stokes) sont caractéristiques de la composition chimique du matériau, de sa structure cristalline ainsi que de ses propriétés électroniques. L'utilisation concerne alors la chimie, l'œnologie, la bijouterie... C'est une des rares méthodes qui permet, à température ambiante, d'obtenir une caractérisation vibrationnelle ou chimique d'un objet. De plus, elle est non destructive et nécessite une très petite quantité de matériau. Dans certains cas particuliers, il est également possible d'estimer des concentrations relatives à l'aide d'une référence connue.
Son inconvénient majeur est la faible intensité de ses raies qui est très inférieure à celle des raies Rayleigh. Il existe cependant un effet Raman en résonance qui fait appel à la théorie vibronique, ce phénomène s'explique plus simplement par le fait que lorsque la fréquence de l'excitatrice est proche des fréquences de transition électronique des atomes il y a un accroissement de l'intensité observée. Cet effet peut être recherché afin de réaliser de meilleures observations.
Degrés de liberté
Pour un composé chimique quelconque, il y a un total de 3N degrés de liberté, où N représente le nombre d'atomes dans le composé. Ce nombre illustre le fait que chaque atome dans une molécule peut bouger dans trois directions différentes: x,y et z[3]. Cependant, lorsqu'on parle de molécules, il est plus commun de considérer le mouvement de toute la molécule au lieu du mouvement de chacun de ses atomes. Ainsi, les 3N degrés de liberté deviennent des mouvements de translation, de rotation et de vibration de la molécule; c'est-à-dire qu'on regarde la molécule selon ces trois degrés. De la même façon, ces trois degrés correspondent aux mouvements de rotation de la molécule selon les axes , , et . Les molécules linéaires ont seulement deux rotations puisque les rotations selon l'axe de liaison ne change pas la position des atomes dans la molécule; il s'agit de la symétrie de la molécule. Les degrés de liberté restants correspondent aux vibrations des modes de la molécule. Ces modes incluent les mouvements d'étirement et de fléxion des liens chimiques de la molécule. Pour une molécule linéaire, le nombre de modes vibrationnels est décrit par:




alors que pour une molécule non linéaire le nombre de modes vibrationnels est décrit par:

Vibrations moléculaires et radiation infrarouge
Les fréquences des vibrations moléculaires vont de moins de 1012 à environ 1014 Hz. Ces fréquences correspondent à la radiation infrarouge (IR) dans la région du spectre électromagnétique. À n'importe quel moment, chaque molécule dans un échantillon a une certaine énergie vibrationnelle. Cependant, cette quantité d'énergie change continuellement à cause des collisions et des interactions avec d'autres molécules dans l'échantillon.
À température ambiante, la plupart des molécules seront dans le plus petit niveau d'énergie, communément appelé l'état fondamental. Quelques molécules seront à des niveaux d'énergies plus grands qui représentent les états excités. Une partie des molécules qui occupent un certain mode vibrationnel peut être calculée grâce à la distribution de Boltzmann. Ces calculs illustrent qu'à des températures relativement petites comme celles utilisées pour faire de la spectroscopie, la plupart des molécules occupent l'état fondamental vibrationnel. Dans ce cas, la molécule peut être excitée à un niveau vibrationnel plus élevé grâce à l'absorption directe d'un photon d'énergie assez grande pour permettre cette transition. En fait, il s'agit du mécanisme de la spectroscopie infrarouge: la radiation infrarouge passe à travers l'échantillon et l'intensité de la lumière transmise est comparée à celle de la lumière incidente. Une réduction d'intensité à une longueur d'onde donnée illustre une absorption d'énergie par un mode vibrationnel. L'énergie d'un photon est représentée par l'équation suivante:

,

où est la constante de Planck et représente la fréquence de la radiation. Ainsi, l'énergie requise pour une telle transition peut être calculée si la fréquence de la radiation incidente est connue.


Distinction de la fluorescence
L'effet Raman est différent de celui de la fluorescence puisqu'il s'agit d'un processus de diffusion. Dans la fluorescence, la lumière incidente est complètement absorbée, ce qui propulse le système dans un état excité. Après un certain temps de vie, le système se désexcite à des niveaux d'énergies inférieurs grâce à l'émission de photons. Essentiellement, le résultat des deux processus est le même: un photon avec une fréquence différente de celle du photon incident est émis et la molécule est amenée à un niveau d'énergie plus faible. Cependant, la différence est que la diffusion Raman peut se produire à n'importe quelle fréquence de la lumière incidente. Contrairement à la fluorescence, la diffusion Raman n'est pas un effet résonant sauf dans le cas de la spectroscopie Raman résonante (en). Concrètement, cela signifie qu'un pic de fluorescence est relié à une fréquence spécifique, alors qu'un pic Raman est différent de la fréquence d'excitation.
Règles de sélection
Une transition Raman d'un état à un autre est permise seulement si la polarisabilité moléculaire de ces états est différente. Pour une vibration, cela implique que la dérivation de la polarisabilité en fonction de la coordonnée associée à la vibration est non nulle : . En général, un mode est activé par effet Raman s'il se transforme selon la même symétrie que celle des formes quadratiques () qui peuvent être vérifiées avec la table de caractères du groupe de symétrie de la molécule en question.


Les règles de sélection spécifient que les transitions de rotation permises sont , où représente l'état de rotation (moment angulaire total). Les transitions de vibration permises sont décrites par , où est l'état vibrationnel.



Application à la spectroscopie Raman
La spectroscopie Raman, ou spectrométrie Raman, est une méthode non destructive permettant de caractériser la composition moléculaire et la structure d'un matériau. Cette technique est complémentaire de la spectroscopie infrarouge qui permet également d'étudier les modes vibrationnels d'un matériau. Cependant les règles de sélection pour les deux spectroscopies peuvent être différentes selon la symétrie de la molécule ou du cristal, de sorte que certains modes vibrationnels peuvent être observés au Raman seulement et d'autres à l'infrarouge seulement.
La méthode consiste à focaliser (avec une lentille) un faisceau de lumière monochromatique (un faisceau laser) sur l'échantillon à étudier et à analyser la lumière diffusée. Cette lumière est recueillie à l'aide d'une autre lentille et envoyée dans un monochromateur et son intensité est alors mesurée avec un détecteur (monocanal type photomultiplicateur ou CPM, multicanal type CCD).
Références
- (de) Adolf Smekal, « Zur Quantentheorie der Dispersion », Naturwissenschaften, vol. 11, no 43, , p. 873–875 (ISSN 0028-1042 et 1432-1904, DOI 10.1007/bf01576902, lire en ligne, consulté le )
- (de) K. G. E, « Der Smekal-Raman-Effekt », Nature, vol. 128, no 3242, , p. 1026–1026 (DOI 10.1038/1281026c0, lire en ligne).
- Laidler, Keith J. (Keith James), 1916-2003., Physical chemistry, Benjamin/Cummings Pub. Co, (ISBN 0805356827, OCLC 8112942, lire en ligne)
Voir aussi
Articles connexes
Liens externes
- La spectroscopie Raman sur le site de l'université de Lyon
- La spectroscopie Raman sur le site du Conseil national de recherches du Canada
 Portail de la physique
Portail de la physique  Portail de l’optique
Portail de l’optique  Portail des sciences quantiques
Portail des sciences quantiques