Protéine Forkhead-P2
La protéine Forkhead-Box P2 (FOXP2) est un facteur de transcription appartenant au groupe des protéines Forkhead-Box[3],[4]. Elle a été découverte pour la première fois en 1998 dans une enquête sur une famille londonienne, parmi laquelle de nombreux membres avaient de profondes difficultés d'élocution se rapprochant de l'aphasie de Broca. Depuis, il a été reconnu que FOXP2 joue un grand rôle dans la transmission du langage, y compris les capacités grammaticales[5].
| FOXP2 | ||
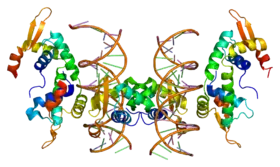 Modèle en bandes de la tête d'une protéine FOXP2 complexée avec un ADN, selon PDB 2A07 | ||
| Caractéristiques générales | ||
|---|---|---|
| Nom approuvé | Facteur de transcription à forkhead 2 | |
| Symbole | FOXP2 | |
| Synonymes | Protéine du langage | |
| Distribution | Vertébrés[1] | |
| 'Homo sapiens' | ||
| Chromosome et locus | 7q31 | |

Dans les médias de masse, le nom de « gène de la parole » a été popularisé pour le gène FOXP2. Mais beaucoup d'autres vertébrés possèdent également ce gène, et FOXP2 semble jouer chez eux un rôle dans les communications verbales. L'inactivation de ce gène, par exemple chez la souris (souris knock-out), ou chez l'homme par mutation, provoque des effets pléiotropes, c'est-à-dire que plusieurs caractères phénotypiques sont modifiés[6].
Structure
C'est le gène FOXP2 qui code la protéine FOXP2 (voir code génétique).
Gène FOXP2
On estime actuellement que le gène FOXP2 occupe au moins 280 kb (280 000 paires de bases) sur le chromosome 7[7]. D'autres publications de 2007 indiquent un nombre de paires de 603 kb[8]. Indépendamment de cela, une très grande proportion de paires de bases constitue des introns, c'est-à-dire des parties non fonctionnelles pour la synthèse de la protéine. 17 exons codants la protéine ont été localisés[9].
La proportion des 2 145 (= 715 × 3) paires de bases sur les 280 000 en tout est, avec 0,6 %, très faible, mais pas extraordinaire. En général, ce rapport varie beaucoup de gène à gène. Il y a par exemple des gènes sans introns, tandis que d'autres dépassent les 95 % d'introns[10].
Protéine FOXP2
La protéine FOXP2 codée par le gène FOXP2 consiste en 715 acides aminés. Elle consiste en 4 domaines principaux :
- une région riche en polyglutamine, consistant en deux domaines voisins de polyglutamine, codés par des séquences répétitives de CAG et CAA[11],
- un domaine en doigt de zinc,
- un domaine en bZIP (glissière à leucine ou leucine zipper),
- un domaine Forkhead, formé par les acides aminés 508 à 584[12].
Le domaine Forkhead se lie à l'ADN. Les domaines en doigt de zinc et bZIP sont importants pour les interactions entre protéines, et participent aussi à la liaison à l’ADN[13].
Fonction
Selon les estimations, la protéine FOXP2 contrôle en tant que facteur de transcription jusqu'à 1000 autres gènes, mais on n'en connait qu'une petite partie. Mais on a des connaissances sûres sur les conséquences d'une malformation de FOXP2.
On trouve la protéine FOXP2 déjà dans l'embryon. Elle s'exprime avant tout dans les régions qui se développeront ultérieurement en cervelet, en thalamus et en ganglions de la base. Le cervelet et les ganglions de la base jouent un grand rôle dans l'apprentissage des capacités motrices complexes, telles que le langage (une capacité nécessitant un long apprentissage)[14].
Troubles du langage et de la parole
La protéine FOXP2 joue un rôle central dans le développement des capacités de langage et de parole. C'est pourquoi des mutations dans le gène et une perte résultante des propriétés de la protéine conduit chez l’homme à des perturbations spécifiques du langage et de la parole, en particulier en ce qui concerne l'articulation et la compréhension du langage[15]. Une série de perturbations connues du langage et de la parole, ainsi que l'autisme, sont pour cela rapportés au domaine du gène FOXP2 sur le chromosome 7[16],[17].
Schizophrénie
Les perturbations du langage sont un des symptômes majeurs de la schizophrénie. C'est pourquoi, immédiatement après la découverte du gène FOXP2, ce gène a été soupçonné de jouer un rôle prédisposant à la survenue de la schizophrénie. Dans une étude comparative, 186 patients schizophrènes (selon le DSM-IV, ils entendent des voix), et 160 sujets de test sains ont été examinés. Ces examens portaient spécialement sur le polymorphisme génétique du gène FOXP2. On a pu établir des différences significatives sur le plan statistique entre les deux groupes, tant en ce qui concerne le génotype (probabilité d'égalité P = 0,007) et la fréquence des allèles (P = 0,0027). On n'a trouvé dans le groupe témoin qu'un seul cas de polymorphisme (rs2396753). Le résultat permet de conclure que le gène FOXP2 peut avoir une influence sur la formation d'une schizophrénie[18].
Découverte


En 1990, des généticiens britanniques de l’Institute of Child Health étudient une anomalie héréditaire du langage qui concerne trois générations d'une famille. Environ la moitié des 30 membres de la famille avait des problèmes sévères en grammaire, syntaxe et vocabulaire[20]. Dans la littérature scientifique, ce groupe est nommé KE-family (famille KE). Elle habite le sud de Londres, et est d'origine pakistanaise. Le généticien Anthony Monaco de l’université d'Oxford a découvert en 1998, avec son groupe de travail, chez les membres de la famille affectés par les troubles du langage un segment du chromosome 7 qu'il a associé au problème de langage de la famille. C'est par des études génétiques sur la famille KE, et sur un jeune (le patient CS), qui n’était pas parent de la famille KE, mais qui montre les mêmes symptômes, que le gène FOXP2, surnommé le « gène de la parole », a été identifié pour la première fois.
La mutation du gène FOXP2 est apparue apparemment chez la grand-mère de la famille. Ses difficultés de langage sont telles que même son mari ne peut déchiffrer ses phrases qu’avec peine. Les trois filles[19] et un des deux fils ont également des difficultés de parole. Parmi les 24 petits-enfants, dix montrent les mêmes symptômes. Les autres membres de la famille du sud de Londres n'ont aucun problème de compréhension[21]. Les troubles manifestés par les membres concernés de la famille KE sont désignés par dyspraxie verbale développementale (Developmental Verbal Dyspraxia, ou en abrégé DVD) – ce qui pourrait se dire en mots courant : incapacité au langage articulé – et rangée sous le code F83 de la CIM-10 (Troubles spécifiques mixtes du développement).
Symptômes d'une mutation de FOXP2
Dyspraxie verbale développementale

Le phénotype général des personnes atteintes de dyspraxie verbale développementale est mis en évidence par le simple test de répétition de mots. Il s'agit d'y répéter, après les avoir entendus, des mots (p.ex. Pardon) et des non-mots (p.ex. Gartin). Les sujets atteints de la mutation y ont pour l’articulation des problèmes significativement plus importants que ceux qui ne sont pas atteints[22]. La difficulté s'accroît progressivement avec la complexité des mots à prononcer.
Dyspraxie orofaciale
Les personnes touchées ont aussi des difficultés à actionner volontairement leurs muscles de la face ; symptôme nommé dyspraxie orofaciale. Ces difficultés ne peuvent pas être attribuées à une incapacité générale de la motricité, parce que les performances motrices des extrémités des patients ne diffèrent pas de celles de personnes normales[22]. La capacité auditive des patients est normalement développée. Le phénotype de la DVD est donc semblable à celui observé chez les patients affligés d'une aphasie de Broca[23]. Cependant, il existe entre les deux maladies des différences importantes du comportement. Par exemple, les aphasiques sont, dans le test de répétition, bien meilleurs pour les mots que pour les non-mots. Les membres de la famille KE touchés par la mutation étaient également mauvais pour les deux catégories. Une explication possible pour cela est que les aphasiques, avant l’apparition de leur maladie, ont appris l'association entre la structure des sons et la signification correspondante des mots. À l'opposé, les membres touchés de la famille KE n'auraient jamais eu la possibilité d'apprendre les structures de l'articulation des mots. C'est pourquoi ils échouent inévitablement en essayant de résoudre le test de répétition de mots en s'appuyant sur la signification des mots[24].
Autres symptômes
Outre les dyspraxies verbale et orofaciale, les membres de la famille KE atteints par la mutation se détachent de leurs parents non atteints par leurs capacités de réception (compréhension du langage) et de production de phrases grammaticalement correctes. Ce déficit comprend l'incapacité de décliner correctement les mots ou de former des phrases établissant des relations simples entre des objets et les descriptions correspondantes. De plus, dans les tests d'intelligence non verbaux les sujets atteints montrent une intelligence significativement plus mauvaise (QI moyen : 86, fourchette : 71 – 111) que les non-atteints (QI moyen : 104, fourchette : 84 – 119). Malgré tout, il y a un large recouvrement entre les deux groupes[22],[24],[25],[26].
Effets des modifications de FOXP2
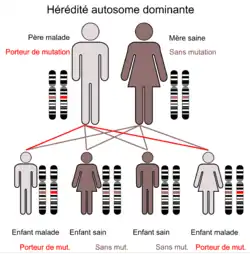

Les perturbations dues à la mutation sont héritées via un gène autosome dominant. Le gène FOXP2 se trouve chez l'homme sur le bras long (q) du chromosome 7 dans la bande 7q31. À l'origine, quand on n’a pu identifier que le chromosome concerné, on l'a appelé « SPCH1 » (de speech, la parole)[19]. En 2006, on a examiné le gène FOXP2 d'un ensemble de 49 sujets souffrant de dyspraxie verbale. Chez l’un des sujets, on a trouvé une mutation non-sens héritée de la mère, et chez deux sujets, une probable mutation faux-sens[12]. Ces résultats permettent de conclure que les mutations du gène FOXP2 sont une cause relativement rare des perturbations du langage et de la parole[2].
Exon 14 (famille KE)
Les membres de la famille KE atteints par cette maladie héréditaire ont une mutation ponctuelle faux-sens dans l’exon 14 du gène. Le nucléotide guanine est remplacé par une adénine. Ceci entraine qu'à la place de l’acide aminé arginine (Arg ou R), en position 553 sur la protéine FOXP2, il y ait une histidine (His ou H). La mutation porte donc la désignation R553H. Cette protéine, à la suite du changement d'acide aminé, ne peut plus assumer ses fonctions.
Au moyen d'imagerie du cerveau des membres de la famille KE, on a constaté des anomalies dans le noyau caudé, une partie des ganglions de la base. Les premiers examens des bases neurologiques ont pu être faits au moyen d'IRMf. Les membres atteints de la famille KE ont montré des déficits structurels bilatéraux. Ceux-ci consistaient avant tout en une épaisseur anormalement faible de substance grise dans la région du noyau caudé des ganglions de la base[22],[26], de la partie antérieure du cervelet[27] et de l'aire de Broca[24]. Cependant, on a trouvé chez ces patients une densité anormalement élevée de substance grise dans le putamen et dans l’aire de Wernicke. De façon intéressante, le volume du noyau caudé est très bien corrélé avec les performances montrées dans le test de langage[22]. Ceci est un indice de l’influence du noyau caudé sur la pathologie de la dyspraxie verbale développementale (DVD).
Les ganglions de la base jouent un rôle décisif dans la planification et le séquencement des mouvements[28]. Des anomalies structurelles dans les régions du striatum (noyau caudé et putamen) signifient donc en général une dégradation du contrôle de la motricité orofaciale (notamment de la bouche). Mais on ne comprend pas pourquoi c'est spécifiquement la motricité de la bouche qui est dégradée, sans que d'autres fonctions motrices ne soient touchées[24].
Exon 7 (mutation non-sens)
En 2005, on a découvert chez des enfants qui n'appartiennent pas à la famille KE, mais qui souffraient aussi de dyspraxie verbale, également dans le gène FOXP2 une mutation non-sens. Ce type de mutation est une mutation non interprétable, par laquelle un codon stop survient, c'est-à-dire un triplet de nucléotides qui provoque l'arrêt de la synthèse de la protéine à cet endroit. Les déficits langagiers et verbaux sont dus dans ce cas aussi à la mutation non-sens[12].
Rupture entre les exons 3 et 4
Un patient, connu dans la littérature comme patient CS a une translocation équilibrée entre un exemplaire du chromosome 5 et un du chromosome 7 : t(5;7)(q22;q31.2). Le point de rupture sur le chromosome 7 est dans le gène FOXP2, entre les exons 3b et 4, et concerne donc toutes les isoformes connues de la protéine FOXP2[2]. Ce patient souffre aussi de symptômes voisins de ceux des membres atteints de la famille KE[29].
Cassure chromosomique
En 2006, on a constaté chez une fillette canadienne une perte (délétion) de parties du chromosome 7 dans les bandes q31 et q32. Dans la région perdue figure aussi le gène FOXP2. L'enfant a de lourdes difficultés de communication sous forme de dyspraxie orofaciale, une difformité visible et un retard dans son développement général. Elle ne peut pas tousser, éternuer ou rire spontanément[30].
Chez la souris

Chez les souris, on a pu désactiver les exons 12 et 13 de FOXP2 (Knock-out). Quand les deux exemplaires du gène sont interrompus, ceci conduit à de lourdes pertes de motricité, une mort prématurée et une incapacité de correspondre par ultrasons. Cette dernière communication se déclenche normalement quand les petits sont éloignés de leur mère. Mais si un seul des deux gènes FOXP2 est interrompu, ceci n'amène qu'un léger retard de développement et un changement important de la communication par ultrasons. On a pu constater sur ces animaux des modifications anormales du cervelet, spécialement des cellules de Purkinje du cortex cérébelleux[31],[32].
En implantant la variante humaine du gène dans le génome des souris, celles-ci ont montré une amélioration importante de leur capacité d'apprentissage par rapport aux souris non implantées. Et on a pu constater des changements dans les ganglions de la base des souris modifiées[33],[34].
Chez le diamant mandarin

L'apprentissage du langage n’est pas limité à l’espèce humaine. Certaines espèces d'animaux, parmi lesquelles les baleines, les chauves-souris, et des oiseaux de trois ordres peuvent apprendre leurs communications acoustiques (« langage animal ») par l'imitation[24]. Les oiseaux chanteurs communiquent par un chant qu'il leur faut largement apprendre. Ils héritent leurs mélodies en imitant les représentants plus âgés de leur espèce. Des petits, élevés isolés de leurs congénères, restent donc muets[14]. Les oiseaux chanteurs sont donc des modèles animaux appropriés pour l’étude de l'apprentissage du langage et de ses prédispositions génétiques. Chez beaucoup d'autres espèces animales, les sons émis sont innés. Même chez les singes, on suppose que leur répertoire de sons est inné[24].
Le chant des diamants mandarins (Taeniopygia guttata) consiste en syllabes différentes, qui donnent des suites structurées. Chez l’homme, la partie importante du cerveau pour l'acquisition du langage est située dans les ganglions de la base. Chez les oiseaux, cette région est nommée aire X. L'expression du gène FOXP2 dans l'aire X culmine pendant la phase d'apprentissage du chant chez les diamants mandarins. Chez les canaris, par contre, FOXP2 est exprimé de façon saisonnière. Dans les phases où le chant est modifié, il est exprimé particulièrement fort. Chez les espèces d'oiseaux qui n'apprennent pas leur chant, comme le pigeon ramier, on n'a pas pu mettre en évidence de telles modifications de l’expression de FOXP2[35].
Au moyen de l'ARN interférent, des chercheurs de l'Institut Max-Planck de génétique moléculaire, à Berlin-Dahlem ont inactivé le gène FOXP2 dans l'aire X de diamants mandarins. Ce procédé consiste à introduire de courtes séquences complémentaires d'ARN dans la cellule, où elles se fixent sur des molécules d'ARNm, et empêchent la production de la protéine FOXP2. Les diamants mandarins chez lesquels on a inactivé FOXP2 imitaient les syllabes de leurs congénères aînés de façon moins précise, et oubliaient des syllabes entières en chantant.
Le mécanisme d'action précis de FOXP2 n'est pas encore connu[36]. En principe le défaut du gène peut altérer les fonctions motrices, par exemple de la syrinx, ou l’enregistrement des chants à apprendre[37],[38].
Évolution moléculaire de FOXP2
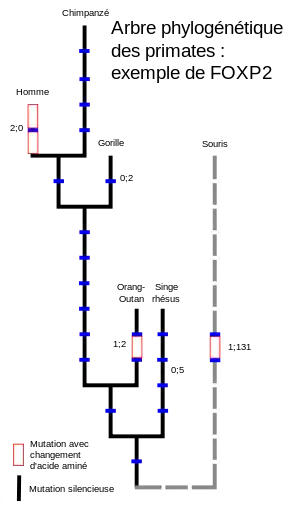
FOXP2 chez les mammifères et autres vertébrés
La protéine FOXP2 chez les mammifères appartient au groupe des protéines fortement conservées : elle ne diffère chez les diverses espèces que très légèrement[11]. Les diverses familles de chauves-souris font cependant exception. On y a trouvé des différences substantielles dans les séquences de FOXP2[39]. Autrement, on trouve des protéines FOXP2 pratiquement identiques chez les oiseaux chanteurs, les poissons et les reptiles[40],[41]. Les segments de gène qui codent la polyglutamine sont généralement connus pour le taux élevé de mutations. Ceci est également le cas pour FOXP2. Tous les taxons étudiés montrent des longueurs variables de polyglutamine. La région de polyglutamine joue un rôle très secondaire pour la fonction de la protéine FOXP2. Si on néglige cette région, la protéine FOXP2 ne diffère de l'orthologue de la souris que par trois acides aminés.
Les lignées évolutives qui ont conduit à l'homme et à la souris se sont séparées il y a environ 40 millions d'années[42],[43]. Le dernier ancêtre commun au chimpanzé et à l'homme a vécu il y a 4,6 à 6,2 millions d'années. Parmi les trois différences d'acides aminés entre l'homme et la souris, l'une est apparue parmi les ancêtres de la souris, aucune entre d'une part la séparation entre lignée des souris et lignée des primates, d'autre part la séparation entre l'homme et le chimpanzé, et deux depuis (voir figure). La protéine FOXP2 de l’orang-outan diffère par deux acides aminés de celle de la souris et par trois de celle de l'homme. Entre les protéines FOXP2 du diamant mandarin et celle de l’homme, il n'y a que sept différences d'acides aminés[11],[44],[45].
La capacité de l’homme à parler repose sur des performances anatomiques et de motricité fine que les autres primates, les plus proches parents de l'homme, ne possèdent pas[46]. Certains groupes de chercheurs supposent que la différence de deux acides aminés entre le chimpanzé et l'homme a conduit au développement du langage chez l'homme[47]. Cette thèse est cependant contestée, parce que d'autres chercheurs n'ont pas pu mettre de correspondance entre les espèces possédant une vocalisation apprise et celles qui possèdent des mutations de ce genre dans FOXP2[40],[41].
Les deux différences avec les plus proches parents de l'homme se trouvent dans l'exon 7. En position 303, une thréonine est changée en asparagine, et en position 325, une asparagine est changée en sérine. Les changements probables dans la structure de la protéine ont été évalués par des calculs de simulation. La mutation en position 325 crée dans la protéine FOXP2 humaine un emplacement potentiellement réactif pour la phosphorylation par la protéine kinase C, avec une légère modification de la structure secondaire de la protéine. On sait, à partir de diverses études, que la phosphorylation des facteurs de transcription avec une structure en forkhead peut être un mécanisme important concernant la régulation de l'expression des gènes[48],[49]. Pour savoir si les deux acides aminés codés dans l'homme sont polymorphes, cet exon a été séquencé chez 44 personnes de divers continents. On n'a trouvé aucun cas de polymorphisme des acides aminés[11].
Le cas des chauves-souris


Tandis que chez la plupart des mammifères on n'a trouvé par des séquençages systématiques d'ADN qu'un taux de mutations très faible sur le gène FOXP2, et portant sur un petit nombre d'acides aminés, il a été signalé chez quelques espèces de chauves-souris des différences substantielles. Les chauves-souris appartiennent au petit nombre de vertébrés qui possèdent la possibilité d'apprendre des sons[50],[51].
L'ordre des chiroptères est divisé en deux sous-ordres : les mégachiroptères et les microchiroptères. Les microchiroptères, insectivores, utilisent l'écholocalisation pour s'orienter et attraper leurs proies. Leurs capacités sensorimotrices sont particulièrement développées. La réception des échos en ultrasons demande une sensibilité auditive, et, selon les espèces, une adaptation orofaciale (bouche) ou nasofaciale (nez)[52]. Les macrochiroptères, frugivores, ne sont pas capables d'écholocalisation.
Le séquençage de l'ADN identifie les exons 7 et 17 comme des domaines où se présente la plus grande variabilité du gène FOXP2. En particulier, il y a de notables différences de structure entre macrochiroptères et microchiroptères. Les données permettent de conclure que les changements dans le gène FOXP2 chez les microchiroptères ont joué un rôle décisif pour le développement de l’écholocalisation[39].
Paléogénétique de FOXP2
Il a d'abord été calculé par la paléogénétique que la variante du gène FOXP2 actuellement diffusée parmi les hommes est vieille de 100 000 à 200 000 ans au plus. Cet intervalle a été calculé par un modèle mathématique fondé spécialement sur les mutations dans les introns. Les introns sont des parties de gène sans influence sur la synthèse des protéines. Comme ils n'ont pas d'influence sur la structure de la protéine, on y observe un taux de mutation bien plus élevé que dans les exons. De ce taux de mutations, on peut reconstruire l'histoire d'un gène[53]. Cet intervalle recouperait convenablement la « naissance » de l'espèce homme donnée par les paléoanthropologues[54],[55]. Du point de vue de l’évolution, ceci est nettement plus tard que la date donnée pour la séparation évolutionnaire entre Homo sapiens et Homo neanderthalensis, il y a 300 000 à 400 000 ans. De ces dates, on peut déduire que le néandertalien ne disposait pas du langage humain[56].
Certains anthropologues soutiennent l’opinion que la rapide diffusion du gène FOXP2 si nécessaire à l'apprentissage du langage renforce la thèse que le langage a été la force motrice de la conquête de la Terre par l'homme[57].
La thèse sur l'âge de la variante actuelle du gène FOXP2 de l'homme, et du fait que le néandertalien ne pouvait pas disposer du langage, a dû être révisée en . On a séquencé le gène FOXP2 extrait d'os de néandertaliens. Et on n'a trouvé aucune différence entre la séquence du néandertalien et celle de l'homme moderne[58],[59].
Le séquençage de l’ADN de trouvailles préhistoriques est un processus très complexe. Les échantillons ne contiennent plus qu'un très faible contenu en ADN endogène. De plus, la contamination des échantillons et des réactifs[60] par de l’ADN humain moderne pose un problème difficile, d'autant plus que l'ADN néandertalien peut différer au plus très peu de l'ADN moderne[61]. On a d'abord analysé l'ADN mitochondrial de deux os néandertaliens différents, qui avaient été trouvés dans la grotte de El Sidrón dans les Asturies, vieux d'environ 43 000 ans. Au moyen de cette analyse on peut établir, par certaines substitutions connues, s'il s'agit d'hommes modernes ou de néandertaliens[62]. Quand il a été établi qu'il s'agissait bien de néandertaliens, on a examiné sur l’exon 7 du gène FOXP2 les deux régions connues pour présenter des mutations depuis la séparation entre l'homme et le chimpanzé. Et on n’a trouvé aucune différence entre la séquence du néandertalien et celle de l'homme moderne. Le néandertalien disposait donc aussi des mutations de FOXP2 rendant possible la parole[59]. La possibilité que cette mutation se soit introduite aussi bien chez Homo sapiens que chez Homo neanderthalensis par croisements entre ces deux groupes est exclue par les résultats des études sur l'ADN mitochondrial[63].
Notes et références
- (de) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en allemand intitulé « Forkhead-Box-Protein P2 » (voir la liste des auteurs).
- (en)« Liste des gènes homologues sur OMA », sur Oma browser (consulté le )
- (en) L. Feuk et coll., « Absence of a Paternally Inherited FOXP2 Gene in Developmental Verbal Dyspraxia. », Am J Hum Genet., vol. 79, , p. 965–72 (PMID 17033973, lire en ligne)
- (en)« FOXP2 », sur OMIM: 605317 (consulté le )
- (en)« Forkhead box protein P2 », sur UniProt: O15409 (consulté le )
- (en) C. Lai et coll., « The SPCH1 region on human 7q31: genomic characterization of the critical interval and localization of translocations associated with speech and language disorder. », Am J Hum Genet, vol. 67/, S., , p. 357–68 (PMID 10880297, lire en ligne)
- (de) P. Schlobinski, Grammatikmodelle : Positionen und Perspektiven, Vandenhoeck & Ruprecht, (ISBN 3-525-26530-1), p. 83–84
- (en)« Gene: FOXP2 », sur Ensembl.org, Ensembl Gene Report for ENSG00000128573 (consulté le ).
- (en) A. F. Wright et N. Hastie, Genes and Common Diseases : Genetics in Modern Medicine., Cambridge (G.-B.), Cambridge University Press, , 544 p. (ISBN 978-0-521-83339-4 et 0-521-83339-6, lire en ligne).
- (en) J. Zhang, D.M. Webb et O. Podlaha, « Accelerated Protein Evolution and Origins of Human-Specific Features: FOXP2 as an Example. », Genetics, vol. 162, , p. 1825–1835 (PMID 12524352, lire en ligne).
- (en) E. H. McConekey, How the Human Genome works, Jones & Bartlett, (ISBN 0-7637-2384-3), p. 5.
- (en) W. Enard, M. Przeworski, S.E. Fisher, C.S. Lai, V. Wiebe, T. Kitano, A.P. Monaco et S. Pääbo, « Molecular evolution of FOXP2, a gene involved in speech and language », Nature, vol. 418, , p. 869–872 (PMID 12192408, DOI 10.1038/nature01025, résumé).
- (en) K. D. MacDermot et coll., « Identification of FOXP2 Truncation as a Novel Cause of Developmental Speech and Language Deficits », Am J Hum Genet, vol. 76/, S., , p. 1074–1080. PMID (PMID 15877281, lire en ligne).
- (de) U. Wahn (dir.), Pädiatrische Allergologie und Immunologie, Elsevier Deutschland, , 985 p. (ISBN 978-3-437-21311-3 et 3-437-21311-3), p. 895.
- (de) S. Haesler, « Also sprach der Zebrafink. », Gehirn&Geist, vol. 12, , p. 52–57
- (en)« Geschwätzige Zebrafinken », Max-Planck-Gesellschaft, (consulté le )
- (en) E.K. O'Brien, X. Zhang, C. Nishimura, J.B. Tomblin et J.C. Murray, « Association of Specific Language Impairment (SLI) to the Region of 7q31. », Am. J. Hum. Genet., vol. 72, , p. 1536–43 (PMID 12721956, lire en ligne)
- (en) T.H. Wassink, J. Piven, V.J. Vieland, J. Pietila, R.J. Goedken, S.E. Folstein et V.C. Sheffield, « Evaluation of FOXP2 as an autism susceptibility gene. », American Journal of Medical Genetics, vol. 114, , p. 566–9 (PMID 12116195, résumé)
- (en) J. Sanjuán, A. Tolosa, J.C. González, EJ. Aguilar, J. Pérez-Tur, C. Nájera, M.D. Moltó et R. de Frutos, « Association between FOXP2 polymorphisms and schizophrenia with auditory hallucinations. », Psychiatr Genet, vol. 16, , p. 67-72 (PMID 16538183, résumé)
- (en) S.E. Fisher, F. Vargha-Khadem, K.E. Watkins, A.P. Monaco et M.E. Pembrey, « Localisation of a gene implicated in a severe speech and language disorder. », Nature Genetics, vol. 16, , p. 67–72 (PMID 16538183, résumé).
- (de) J. Cohen, « Die Evolution der Sprache », Technology Review, vol. 2, .
- Bahnsen et Willmann 2001.
- (en) K.E. Watkins, N.F. Dronkers et F. Vargha-Khadem, « Behavioural analysis of an inherited speech and language disorder: comparison with acquired aphasia », Brain, vol. 125, , p. 452–464 (PMID 11872604).
- (en) A. R. Damasio et N. Geschwind, « The neural basis of language », Annu Rev Neurosci, vol. 7, , p. 127–147.
- (de) S. Haesler, Studien zur Evolution und Funktion des FoxP2-Gens in Singvögeln : Dissertation, FU, Berlin, .
- (en) K.J. Alcock, R.E. Passingham, K.E. Watkins et F. Vargha-Khadem, « Oral dyspraxia in inherited speech and language impairment and acquired dysphasia », Brain Lang, vol. 75, , p. 17–33 (PMID 11023636).
- (en) F. Vargha-Khadem et coll., « Neural basis of an inherited speech and language disorder. In: », Proc Natl Acad Sci U S A, vol. 95, , p. 12695–12700 (PMID 9770548).
- (en) E. Belton, C.H. Salmond, K.E. Watkins, F. Vargha-Khadem et D.G. Gadian, « Bilateral brain abnormalities associated with dominantly inherited verbal and orofacial dyspraxia », Hum Brain Mapp, vol. 18, , p. 194–200 (PMID 12599277).
- (en) A.M. Graybiel, « Building action repertoires: memory and learning functions of the basal ganglia », Curr Opin Neurobiol, vol. 5, , p. 733–741 (PMID 8805417).
- (de) Peter Markl, « Von Menschen und Zebrafinken », Wiener Zeitung, (lire en ligne).
- (en) S. Zeesman, M.J. Nowaczyk, I. Teshima, W. Roberts, J.O. Cardy, J. Brian, L. Senman, L. Feuk, L.R. Osborne et S.W. Scherer, « Speech and language impairment and oromotor dyspraxia due to deletion of 7q31 that involves FOXP2 », Am J Med Genet A, vol. 140, , p. 509–514 (PMID 16470794).
- (en) W. Shu et coll., « Altered ultrasonic vocalization in mice with a disruption in the Foxp2 gene », Proc Natl Acad Sci U S A, vol. 102, , p. 9643–9648 (PMID 15983371, lire en ligne).
- (en) E. Fujita, Y. Tanabe, A. Shiota, M. Ueda, K. Suwa, M.Y. Momoi et T. Momoi, « Ultrasonic vocalization impairment of Foxp2 (R552H) knockin mice related to speech-language disorder and abnormality of Purkinje cells », Proc Natl Acad Sci U S A, vol. 105, , p. 3117–3122 (PMID 18287060, lire en ligne).
- (en) C. Schreiweis, E. Burguière, U. Bornschein, S. Goyal, W. Hevers, R. Mundry, W. Enard, A. M. Graybiel, « Humanized Foxp2 alters learning in differently balanced cortico-basal ganglia circuits », Conference Neuroscience 2011 Proceedings, (consulté le ).
- (de) « FOXP2. "Sprachgen" hilft auch beim Lernen », spektrumdirekt, (consulté le ).
- (en) S. Haesler, K. Wada, A. Nshdejan, E.E. Morrisey, T. Lints, E.D. Jarvis et C. Scharff, « FoxP2 expression in avian vocal learners and non-learners », J Neurosci, vol. 24, , p. 3164–3175 (PMID 15056696).
- (en) S.A. White, S.E. Fisher, D.H. Geschwind, C. Scharff et T.E. Holy, « Singing Mice, Songbirds, and More: Models for FOXP2 Function and Dysfunction in Human Speech and Language », The Journal of Neuroscience, vol. 26, , p. 10376–10379 (PMID 17035521).
- (de) « Schlechte Gesangsschüler – Wissenschaftler schalten das Gen FOXP2 bei Zebrafinken stumm und kriegen was zu hören », Max-Planck-Gesellschaft, (consulté le ).
- (en) S. Haesler, C. Rochefort, B. Georgi, P. Licznerski, P. Osten et C. Scharff, « Incomplete and inaccurate vocal imitation after knockdown of FoxP2 in songbird basal ganglia nucleus Area X », PLoS Biol., vol. 5, , e321 (PMID 18052609, lire en ligne).
- (en) G. Li, J. Wang, S.J. Rossiter, G. Jones et S. Zhang, « Accelerated FoxP2 evolution in echolocating bats », PLoS ONE, vol. 2, no 9, , e900 (PMID 17878935, DOI 10.1371/journal.pone.0000900, lire en ligne).
- (en) D.M. Webb et J. Zhang, « FoxP2 in song-learning birds and vocal-learning mammals », J Hered, vol. 96, , p. 212–216 (PMID 15618302).
- (en) C. Scharff et S. Haesler, « An evolutionary perspective on FoxP2: strictly for the birds? », Curr Opin Neurobiol, vol. 15, , p. 694–703 (PMID 16266802).
- (en) S. Kumar, et S. B. Hedges, « A molecular timescale for vertebrate evolution. In: », Nature, vol. 392, , p. 917–920 (PMID 9582070).
- (en) E. Eizirik, W.J. Murphy et S.J. O'Brien, « Molecular dating and biogeography of the early placental mammal radiation », J. Hered., vol. 92, , p. 212–219 (PMID 11396581, lire en ligne).
- (en) I. Teramitsu, L.C. Kudo, S.E. London, D.H. Geschwind et S.A. White, « Parallel FoxP1 and FoxP2 expression in songbird and human brain predicts functional interaction », J Neurosci, vol. 24, , p. 3152–3163 (PMID 15056695).
- (en) S. Haesler, K. Wada, A. Nshdejan, E.E. Morrisey, T. Lints, E.D. Jarvis et C. Scharff, « FoxP2 expression in avian vocal learners and non-learners », J Neurosci, vol. 24, , p. 3164–3175 (PMID 15056696).
- (en) P. Liebermann, The Biology and Evolution of Language, Cambridge (MA), Harvard University Press, (ISBN 0-674-07413-0).
- (en) W. Enard, M. Przeworski, S.E. Fisher, C.S. Lai, V. Wiebe, T. Kitano, A.P. Monaco et S. Pääbo, « Molecular evolution of FOXP2, a gene involved in speech and language. In: », Nature, vol. 418, , p. 869–872 (PMID 12192408).
- (en) G.J. Kops, R.H. Medema, J. Glassford, M.A. Essers, P.F. Dijkers, P.J. Coffer, E.W. Lam et B.M. Burgering, « Control of cell cycle exit and entry by protein kinase b-regulated forkhead transcription factors », Mol. Cell. Biol., vol. 22, , p. 2025–2036 (PMID 11884591).
- (en) A. Brunet, A. Bonni, M.J. Zigmond, M.Z. Lin, P. Juo, L.S. Hu, M.J. Anderson, KC, Arden, J. Blenis et M.E. Greenberg, « Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor », Cell, vol. 96, , p. 857–868 (PMID 10102273).
- (en) J. W. Boughman, « Vocal learning by greater spear-nosed bats », Proc Biol Sci, vol. 265, , p. 227–233 (PMID 9493408, lire en ligne).
- (en) G. Jones et R. D. Ransome, « Echolocation calls of bats are influenced by maternal effects and change over a lifetime. », Proc Biol Sci, vol. 252, , p. 125–128 (PMID 8391702, DOI 10.1098/rspb.1993.0055, résumé).
- (en) C. F. Moss CF, S. R. Sinha: Neurobiology of echolocation in bats. In: Current Opinion in Neurobiology, 13/2003, S. 751–758. (en) C. F. Moss et S. R. Sinha, « Neurobiology of echolocation in bats », Current Opinion in Neurobiology, vol. 13/, S., , p. 751–758. PMID (PMID 14662378, résumé).
- (de) « Paläogenetik: Wie alt ist die Sprache? », Geo Magazin, (consulté le ).
- (de) U. Bahnsen, « Paläogenetik : Gitarrist im Genlabor », Zeit Wissen, vol. 34, (lire en ligne [archive du ]).
- (en) R. G. Klein, The Human Career : Human Biological and Cultural Origins, Chicago (Ill.), University Chicago Press, , 810 p. (ISBN 0-226-43963-1).
- (de) E. D. Jarasch, « Genetische Spuren der Menschwerdung » [archive du ], BIOPRO Baden-Württemberg GmbH, (consulté le ).
- (de) F. Carmine, Genomtechnologie und Stammzellforschung- ein verantwortbares Risiko?, Eschborn (Hesse), Govi-Verlag, (ISBN 3-7741-1000-X), p. 53.
- (en) Mason Inman, « Neandertals Had Same „Language Gene“ as Modern Humans », National Geographic News, (lire en ligne).
- (en) J. Krause et coll., « The derived FOXP2 variant of modern humans was shared with Neandertals », Current Biology, vol. 17, , p. 1908–1912 (PMID 17949978, lire en ligne).
- (en) M. Hofreiter, D. Serre, H.N. Poinar, M. Kuch et S. Pääbo, « Ancient DNA », Nat. Rev. Genet., vol. 2, , p. 353–359 (PMID 11331901, résumé).
- (en) S. Pääbo, « Human evolution », Trends Cell Biol, vol. 9, , M13–6 (PMID 10611673).
- (en) R. E. Green et coll., « Analysis of one million base pairs of Neanderthal DNA », Nature, vol. 444, , p. 330–336 (PMID 17108958, résumé).
- (de) « Neandertaler hat Sprachgen », sur Archaeologie-online.de, (consulté le ) Offline « pour le moment ».
Voir aussi
Textes de recherche
- (en) G. Konopka, J.M. Bomar, K. Winden, G. Coppola, Z.O. Jonsson, F. Gao, S. Peng, T.M. Preuss, J.A. Wohlschlegel et D.H. Geschwind, « Human-specific transcriptional regulation of CNS development genes by FOXP2 », Nature, vol. 462, , p. 213–217 (PMID 19907493)
- (en) C.S. Lai, S.E. Fisher, J.A. Hurst, F. Vargha-Khadem et A.P. Monaco, « A forkhead-domain gene is mutated in a severe speech and language disorder », Nature, vol. 413, , p. 519–523 (PMID 11586359)
- (en) C.S. Lai, S.E. Fisher, J.A. Hurst, F. Vargha-Khadem et A.P. Monaco, « FOXP2 and the Neuroanatomy of Speech and Language », Nature Reviews Neuroscience, vol. 6, , p. 131–138 (PMID 15685218)
- (en) F. Vargha-Khadem, K. Watkins, K. Alcock, P. Fletcher et R. Passingham, « Praxic and nonverbal cognitive deficits in a large family with a genetically transmitted speech and language disorder », Proc Nat Acad Sci, vol. 92, , p. 930–933 (PMID 7846081)
- (en) H. A. Bruce et R. L. Margolis, « FOXP2: novel exons, splice variants, and CAG repeat length stability », Human Genetics, vol. 111, , p. 136–144 (PMID 12189486)
- (en) C.S. Lai, S.E. Fisher, J.A. Hurst, F. Vargha-Khadem et A.P. Monaco, « A novel forkhead-domain gene is mutated in a severe speech and language disorder. In: », Nature, vol. 413, , p. 519–523 (PMID 11586359)
- (en) B. Wang, D. Lin, C. Li et P. Tucker, « Multiple domains define the expression and regulatory properties of Foxp1 forkhead transcriptional repressors », J Biol Chem, vol. 278/, S., , p. 24259–24268. PMID (PMID 12692134, lire en ligne)
- (en) J.C. Stroud, Y. Wu, D.L. Bates, A. Han, K. Nowick, S. Pääbo, H. Tong et L. Chen, « Structure of the forkhead domain of FOXP2 bound to DNA », Structure, vol. 14, , p. 159–166 (PMID 16407075)
Manuels
- (de) W. Bigenzahn et G. Böhme, Sprach-, Sprech-, Stimm- und Schluckstörungen, Elsevier Deutschland, , 481 p. (ISBN 978-3-437-46950-3 et 3-437-46950-9, lire en ligne)
- (en) R. J. McCauley, Assessment of Language Disorders in Children., Mahwah (N. J.), Lawrence Erlbaum Associates, , 364 p. (ISBN 0-8058-2562-2), p. 118
- (de) Stefanie Otte, Gibt es Zusammenhänge zwischen einer expressiven Sprachentwicklungsstörung und einem zentro- temporalen Sharp- Wave Fokus (Rolando- Fokus) mit der weiteren Entwicklung? : Dissertation, Julius-Maximilians Universität zu Würzburg, (lire en ligne), p. 82–83
- (de) Harald Teepe, Welche Bedeutung haben die Neurowissenschaften für die Fremdsprachendidaktik? : Dissertation, RWTH Aachen, , 233 p. (lire en ligne)
Popularisation
- (de) Ulrich Bahnsen et Urs Willmann, « Wie Gene die Lippen spitzen. », Die Zeit, (lire en ligne)
- (de) Ruth Berger, Warum der Mensch spricht : Eine Naturgeschichte der Sprache, Eichborn, , 304 p. (ISBN 978-3-8218-5687-2)
- (de) Sebastian Haesler, « Also sprach der Zebrafink. », Gehirn&Geist, vol. 12, , p. 52–57 (lire en ligne)
- (de) Annette Leßmöllmann, « Raus mit der Sprache! », Zeit Wissen, (lire en ligne)
- (en) Bijal P. Trivedi, « Scientists Identify a Language Gene », National Geographic Today, (lire en ligne)
- (de) « FOXP2 in der RCSB Protein-Datenbank » (consulté le )
- (de) U. Bahnsen, « Sprich oder stirb! », Sonntagszeitung, (lire en ligne)
- (en) A. MacAndrew, « FOXP2 and the Evolution of Language », 2003, m.à j. 2005 (consulté le )
- (de) K. Seefeldt, « Der kleine Unterschied », Telepolis, (consulté le )
Articles connexes
 Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire  Portail de la linguistique
Portail de la linguistique