Maladie de Rendu-Osler
La maladie de Rendu-Osler ou maladie de Rendu-Osler-Weber ou télangiectasie hémorragique familiale (en anglais, hereditary hemorrhagic telangiectasia ou HHT) est une angiomatose de transmission autosomique dominante, faisant partie des phacomatoses. Elle associe des manifestations cutanéo-muqueuses (télangiectasies) et des malformations vasculaires résultant de l'absence de capillaires entre les veines et les artères. Ces malformations se localisent partout avec des risques de rupture faisant la gravité de cette maladie.
Pour les articles homonymes, voir HHT.
| Spécialité | Génétique médicale |
|---|
| CIM-10 | I78.0 |
|---|---|
| CIM-9 | 448.0 |
| OMIM | 187300 600376 601101 610655 175050 |
| DiseasesDB | 9303 |
| MedlinePlus | 000837 |
| eMedicine |
957067 ped/1668 derm/782 |
| MeSH | D013683 |
| GeneReviews | Hereditary Hemorrhagic Telangiectasia |
![]() Mise en garde médicale
Mise en garde médicale
Elle doit son nom aux médecins Henri Rendu et William Osler.
Cause
Cinq formes génétiques de maladie de Rendu-Osler ont été identifiées. Plus de 80 % des cas sont dus à des mutations sur les gènes ENG ou ACVRL1[1], les deux formes ayant une présentation clinique un peu différente[2]. Au total, plus de 600 mutations différentes ont été identifiées[3]. La première mutation a été trouvée sur le gène ENG codant l'endogline[4], une glycoprotéine faisant partie du complexe protéique récepteur du TGFβ.
Manifestations
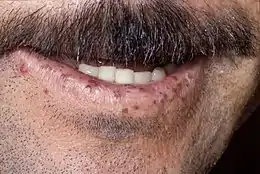
La manifestation la plus commune est une épistaxis (saignements de nez spontanés et fréquents) commençant la plupart de temps vers 12 ans. L'examen montre la présence de télangiectasies (petites taches rouges dues à la dilatation de petits vaisseaux) au niveau des mains, lèvres, conjonctives, paupières et muqueuses nasales.
Des hémorragies digestives surviennent dans un quart des cas après 50 ans. L'atteinte hépatique est courante, surtout chez la femme mais est rarement symptomatique[5]. Présente dans un tiers des cas, elle peut parfois conduire à des complications graves[6] : encéphalopathie hépatique, hypertension portale, insuffisance hépatique aiguë. Les malformations se localisant dans le cerveau, l'appareil digestif ou pulmonaire sont responsables de la mortalité de cette maladie.
Les multiples shunts artério-veineux peuvent conduire à une augmentation importante du débit cardiaque pouvant entraîner une insuffisance cardiaque[7]. La maladie peut également se compliquer d'une hypertension artérielle pulmonaire par occlusion des artérioles pulmonaires[8].
Diagnostic
Le diagnostic de maladie de Rendu-Osler se base sur la présence de malformations artério-veineuses cutanées ou viscérales. Les critères diagnostiques incluent : saignement de nez spontané et récurrent (jusqu'à plus d'une trentaine par semaine). La perte de sang peut atteindre un litre par semaine, nécessitant des transfusions régulières ; télangiectasies multiples et typiquement localisées au niveau des lèvres, de la bouche, du nez et des doigts ; malformations vasculaires pulmonaires, cérébrales, hépatiques, vertébrales et intestinales ; un parent au premier degré atteint de la maladie
Le diagnostic est certain quand trois signes sont présents, probable quand deux signes sont présents et peu probable quand un seul signe est présent[9].
Traitement
La prise en charge de la maladie a fait l'objet de recommandations publiées en 2011[10].
Plusieurs molécules anti-angiogenèse approuvées pour d'autres conditions, comme le cancer, ont été étudiées dans un certain nombre d'essais cliniques chez des patients Rendu-Osler[11]. Le traitement à l'anticorps anti-VEGF bevacizumab, par exemple, a été trouvé être associé à des effets bénéfiques dus notamment à une réduction du nombre d'épisodes d'épistaxis chez les patients traités[12]. Le thalidomide, un autre médicament anti-angiogenèse[13],[14], a également été rapportée pour avoir des effets encourageants chez les patients Rendu-Osler[15]. Une étude a notamment démontré que le thalidomide induit la maturation des vaisseaux dans un modèle de souris de la maladie de Rendu-Osler, et aussi réduit la gravité et la fréquence des saignements de nez chez les patients traités. Les taux d'hémoglobine dans le sang de ces patients traités ont augmenté à la suite d'une baisse des pertes hémorragiques et de la stabilisation de leur vaisseaux sanguins[16].
Le thalidomide pourrait être, une fois cette indication validée officiellement par l'ANSM, le premier médicament à proposer un traitement efficace qui permet d'arrêter les épistaxis et augmenter le taux d'hémoglobine dans le sang[16]. Le thalidomide est toutefois un médicament tératogène et qui ne peut être utilisé qu'avec une contraception efficace chez la patiente en âge de procréer.
Notes et références
- (en) Faughnan ME, Palda VA, Garcia-Tsao G. et al., « International guidelines for the diagnosis and management of hereditary hemorrhagic telangiectasia », J Med Genet., vol. 48, no 2, , p. 73–87 (PMID 19553198, DOI 10.1136/jmg.2009.069013)
- Lesca G, Olivieri C, Burnichon N et al. Genotype-phenotype correlations in hereditary hemorrhagic telangiectasia: data from the French-Italian HHT network, Genet Med, 2007,9:14-22
- (en) HHT Mutation Database (Hereditary Hemorrhagic Telangiectasia)
- (en) McAllister KA, Grogg KM, Johnson DW et al. « Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1 » Nat Genet. 1994;8:345-351
- (en) Garcia-Tsao G, « Liver involvement in hereditary hemorrhagic telangiectasia (HHT) » J Hepatol. 2007;46:499-507.
- (en) Larson AM, « Liver disease in hereditary hemorrhagic telangiectasia » J Clin Gastroenterol. 2003;36:149-58.
- (en) Ginon I, Decullier E, Finet G et al. « Hereditary hemorrhagic telangiectasia, liver vascular malformations and cardiac consequences » Eur J Intern Med. 2013;24:e35-e39.
- (en) Trembath RC, Thomson JR, Machado RD et al. « Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia » N Engl J Med. 2001;345:325-34.
- (en) Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, Kjeldsen AD, Plauchu H, « Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome) » Am J Med Genet. 2000;91:66-7
- Faughnan ME, Palda VA, Garcia-Tsao G et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia, J Med Genet, 2011;48:73-87
- (en) Shovlin CL, « Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment », Blood Rev, vol. 24, , p. 203-19 (PMID 20870325, DOI 10.1016/j.blre.2010.07.001)
- Dupuis-Girod S, Ginon I, Saurin JC, Marion D, Guillot E, Decullier E, Roux A, Carette MF, Gilbert-Dussardier B, Hatron PY, Lacombe P, Lorcerie B, Rivière S, Corre R, Giraud S, Bailly S, Paintaud G, Ternant D, Valette PJ, Plauchu H, Faure F, « Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output », JAMA, vol. 307, , p. 948-55 (PMID 22396517, DOI 10.1001/jama.2012.250)
- D'Amato RJ, Loughnan MS, Flynn E, Folkman J, « Thalidomide is an inhibitor of angiogenesis », Proc Natl Acad Sci U S A, vol. 91, , p. 4082-5 (PMID 7513432, DOI 10.1073/pnas.91.9.4082)
- (en) Therapontos C, Erskine L, Gardner ER, Figg WD, Vargesson N, « Thalidomide induces limb defects by preventing angiogenic outgrowth during early limb formation », Proc Natl Acad Sci USA, vol. 106, , p. 8573-8 (PMID 19433787, DOI 10.1073/pnas.0901505106)
- (en) Franchini M, Frattini F, Crestani S, Bonfanti C, « Novel treatments for epistaxis in hereditary hemorrhagic telangiectasia: a systematic review of the clinical experience with thalidomide », J Thromb Thrombolysis, (PMID 23143669)
- (en) Lebrin F, Srun S, Raymond K, Martin S, van den Brink S, Freitas C, Bréant C, Mathivet T, Larrivée B, Thomas JL, Arthur HM, Westermann CJ, Disch F, Mager JJ, Snijder RJ, Eichmann A, Mummery CL, « Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia », Nat Med, vol. 16, , p. 420-8 (PMID 20364125, DOI 10.1038/nm.2131)
Voir aussi
Bibliographie
- (en) Alan E Guttmacher, Jamie McDonald, Hereditary Hemorrhagic Telangiectasia in GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2006 www.genetests.org