Orbitale moléculaire
En chimie quantique, une orbitale moléculaire est une fonction mathématique décrivant le comportement ondulatoire d'un électron dans une molécule. Cette fonction peut être utilisée pour calculer la configuration électronique des molécules, la distribution spatiale et énergétique des électrons dans les molécules, et en déduire des propriétés physiques ou chimiques, comme la probabilité de trouver un électron dans une région donnée. La combinaison linéaire d'orbitales atomiques offre un moyen simple de construire une représentation approchée des orbitales moléculaires, notamment pour les descriptions qualitatives. Cette méthode est très utilisée par exemple pour établir un modèle simple des liaisons chimiques dans les molécules, décrites à l'aide de la théorie de l'orbitale moléculaire. La méthode de Hartree-Fock et l'hybridation permettent également d'y parvenir. Les méthodes plus actuelles de la chimie numérique calculent les orbitales moléculaires de chaque électron placé dans le champ électrique généré par l'ensemble des noyaux atomiques et la distribution spatiale moyenne des autres électrons. Lorsque deux électrons occupent la même orbitale, le principe d'exclusion de Pauli impose qu'ils soient de spin opposé.
Pour les articles homonymes, voir orbitale.
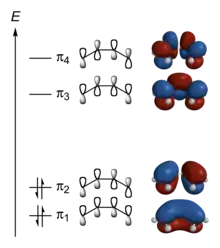
Les orbitales moléculaires dérivent des interactions entre orbitales atomiques permises lorsque les symétries (au sens de la théorie des groupes) de ces dernières sont compatibles entre elles. L'efficacité des interactions entre orbitales atomiques est déterminée par leur recouvrement, qui est important si ces orbitales sont d'énergie voisine. Le nombre d'orbitales moléculaires ainsi formées dans une molécule doit être égal au nombre total des orbitales atomiques des atomes constituant cette molécule.
L'essentiel des caractéristiques et des manières de représenter les orbitales atomiques sont transposables aux orbitales moléculaires. L'étude de ces dernières est généralement plus compliquée car aucune résolution analytique n'est possible ; il est donc nécessaire de passer par des approximations pour les déterminer. Les descriptions les plus précises des fonctions d'onde des électrons dans les molécules, comme les méthodes post-Hartee-Fock telles que l'interaction de configuration, ne modélisent d'ailleurs pas d'orbitales moléculaires.
Combinaison linéaire d'orbitales atomiques
Le concept d'orbitales moléculaires a été introduit par Friedrich Hund[1],[2],[3] et Robert Mulliken[4],[5] dans les années 1927-1928. La combinaison linéaire d'orbitales atomiques, ou LCAO, a été introduite en 1929 par John Lennard-Jones[6] en montrant comment déduire la configuration électronique des molécules de fluor F2 et d'oxygène O2 à partir de principes de la mécanique quantique. Mathématiquement, il s'agit de développer l'orbitale moléculaire sur une base d'orbitales atomiques, ou une série d'orbitales atomiques que l'on supposera être proche d'une base. Cette approche qualitative de la théorie de l'orbitale moléculaire est l'une des sources de la chimie quantique actuelle. La combinaison linéaire d'orbitales atomiques peut ainsi permettre de modéliser les orbitales moléculaires qui se forment entre les atomes constituant des molécules. Comme dans le cas des atomes, il est possible de faire intervenir une équation de Schrödinger pour décrire de telles orbitales. Plus précisément, cette méthode fournit des solutions approchées des équations de Hartree-Fock correspondant à l'approximation des particules indépendantes de l'équation de Schrödinger pour la molécule. Dans le cas des molécules diatomiques, les fonctions d'onde obtenues sont représentées mathématiquement par les équations :


où et sont les fonctions d'onde de l'orbitale moléculaire liante et de l'orbitale moléculaire antiliante respectivement, et sont les fonctions d'onde des atomes et , et et sont des coefficients ajustables. Ces coefficients peuvent être positifs ou négatifs selon l'énergie et la symétrie des orbitales atomiques. Lorsque les deux atomes se rapprochent, leurs orbitales atomiques se recouvrent en formant des régions où la densité électronique est élevée, ce qui conduit à la formation d'orbitales moléculaires entre les atomes. Les atomes sont ainsi liés par l'attraction coulombienne entre les noyaux, qui sont chargés positivement, et les électrons des orbitales liantes, qui sont chargés négativement.








Diagrammes d'orbitales moléculaires
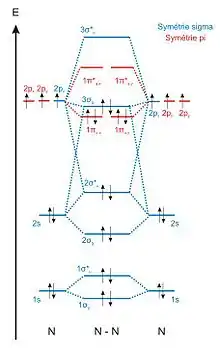
L'approche qualitative de l'analyse des orbitales moléculaires utilise les diagrammes d'orbitales moléculaires afin de visualiser les interactions à l'origine des liaisons chimiques entre atomes dans les molécules. Dans ce type de diagrammes, les orbitales moléculaires sont représentées par des traits horizontaux situés d'autant plus haut que l'énergie correspondante est élevée. Les orbitales dégénérées sont représentées par des traits adjacents séparés par un espace vide et situés au même niveau. Les électrons des orbitales moléculaires sont ensuite distribués sur ces différents traits en satisfaisant à la fois la règle de Hund et le principe d'exclusion de Pauli, c'est-à-dire en remplissant les niveaux d'énergie d'abord par des électrons célibataires sur chaque orbitale avant de les apparier avec un électron de spin opposé (règle de Hund), à raison d'au plus deux électrons sur chaque orbitale (principe d'exclusion de Pauli).
Quelques propriétés fondamentales des diagrammes d'orbitales moléculaires :
- les orbitales de base comprennent les orbitales atomiques permettant la formation des orbitales moléculaires, qui peuvent être liante ou antiliantes ;
- le nombre d'orbitales moléculaire est égal au nombre d'orbitales atomiques du développement linéaire de la base
- lorsque la molécule présente certaines symétries, les orbitales atomiques dégénérées, c'est-à-dire qui présentent la même énergie, sont regroupées en combinaisons linéaires (appelée SO, de l'anglais symmetry adapted atomic orbitals) qui appartient à la représentation d'un groupe symétrique, de sorte que les fonctions d'onde qui décrivent ce groupe sont appelées symmetry-adapted linear combinations (SALC) ;
- le nombre d'orbitales moléculaires appartenant à un groupe est égal au nombre des orbitales atomiques adaptées à cette symétrie et appartenant à cette représentation ;
- à l'intérieur d'une représentation particulière, les orbitales atomiques adaptées à la symétrie se mélangent d'autant plus que leur niveau d'énergie atomique est plus proche.
Typologie des orbitales moléculaires
Orbitales liantes, antiliantes, non liantes

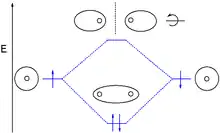
Une orbitale moléculaire est dite liante lorsque les orbitales atomiques dont elle résulte sont en phase : l'interférence de leurs fonctions d'onde est constructive. À l'inverse, une orbitale moléculaire est dite antiliante lorsque les orbitales atomiques dont elle résulte sont en opposition de phase : l'interférence de leurs fonctions d'onde est destructive, et il existe un plan nodal entre les deux atomes, dans lequel la fonction d'onde de l'orbitale moléculaire est d'amplitude nulle. Enfin, une orbitale moléculaire est dite non liante lorsque les orbitales atomiques dont elle résulte sont dépourvues de symétries compatibles leur permettant d'interagir.
 Orbitale liante de la molécule d'hydrogène
Orbitale liante de la molécule d'hydrogène
Les orbitales liantes ont toujours un niveau d'énergie inférieur à ceux des orbitales atomiques dont elles sont issues, ce qui explique qu'un doublet liant soit toujours plus stable que les orbitales atomiques individuelles. À l'inverse, les doublet antiliants ont une énergie plus élevée que les orbitales atomiques dont ils proviennent, ce qui explique leur action répulsive entre leurs atomes d'origine. Enfin, les doublets non liants ont un niveau d'énergie égal à celui de l'une des orbitales atomiques dont ils proviennent, ce qui explique leur absence d'effet liant ou antiliant.
Qualitativement, la densité de probabilité de présence des électrons d'une orbitale liante est maximum entre les noyaux atomiques — mais pas nécessairement sur l'axe internucléaire — ce qui tend à attirer les atomes l'un vers l'autre par attraction coulombienne, d'où l'effet attractif de ce type liaison. Dans le cas d'une orbitale antiliante, en revanche, la densité de probabilité de présence des électrons est maximum derrière chaque noyau atomique, ce qui tend à éloigner les atomes l'un de l'autre pour la même raison, d'où l'effet répulsif de ce type de liaison. Dans le cas d'une orbitale non liante, la géométrie de la densité de probabilité de présence des électrons est sans effet sur l'attraction ou la répulsion des atomes l'un par rapport à l'autre.
Orbitales σ, π, δ et φ
Les types d'interactions entre orbitales atomiques peuvent être classés en fonction de la symétrie des orbitales moléculaires. Ces symétries sont dites σ (sigma), π (pi), δ (delta) ou φ (phi), et reflètent les symétries observées pour les orbitales atomiques s, p, d et f. Il est possible en théorie de poursuivre, par exemple avec la symétrie γ (gamma), correspondant aux orbitales atomiques g. Le nombre de plans nodaux contenant les deux atomes de la liaison vaut 0 pour les orbitales moléculaires σ, 1 pour les orbitales π, 2 pour les orbitales δ, etc.
- Une liaison σ résulte de la combinaison de deux orbitales atomiques s ou de deux orbitales pz. Elle s'étend le long de l'axe passant par les deux noyaux atomiques, avec une symétrie axiale autour de cet axe. L'orbitale σ* antiliante présente également une symétrie axiale autour de l'axe internucléaire, mais avec un plan nodal orthogonal à cet axe entre les deux atomes liés[7].

- Une liaison π résulte de la combinaison d'orbitales atomiques px ou py. Une rotation d'une telle orbitale autour de l'axe internucléaire introduit un déphasage. Les orbitales réelles présentent un plan nodal passant par les deux noyaux atomiques. Une orbitale π* antiliante présente, outre le plan nodal comprenant l'axe internucléaire, un second plan nodal perpendiculaire au premier et passant entre les deux atomes[7],[8],[9],[10].
- Une liaison δ résulte de la combinaison d'orbitales atomiques dxy ou dx2–y2. On observe de telles orbitales dans le complexes de métaux de transition. Une orbitale de symétrie δ présente deux plans nodaux contenant les deux noyaux atomiques, tandis qu'une orbitale δ* antiliante présente en plus un plan nodal passant entre les deux noyaux.
- Une liaison φ résulte de la combinaison d'orbitales atomiques f ; la molécule de diuranium U2 est l'une des seules espèces chimiques connues présentant une telle liaison[11].
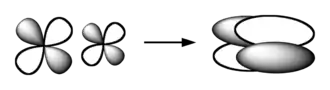

Symétrie gerade et ungerade
Les molécules centrosymétriques, c'est-à-dire qui présentent un centre d'inversion, sont par exemple les molécules diatomiques homonucléaires X2, les molécules à géométrie carrée plane EX4 et les molécules à géométrie octaédrique EX6, mais pas les molécules diatomiques hétéronucléaires XY et les molécules à géométrie tétraédrique EX4, par exemple.
Lorsque l'inversion par rapport au centre de symétrie d'une molécule centrosymétrique ne change pas la phase d'électron de l'orbitale, celle-ci est dite gerade, noté g, d'un mot allemand signifiant « pair » ; si l'inversion par rapport au centre de symétrie conduit à un changement de phase de l'orbitale, alors celle-ci est dite ungerade, noté u, d'un mot allemand signifiant « impair ».
Une orbitale liante de symétrie σ est ainsi σg, car construite par addition d'orbitales atomiques s, ce qui est symétrique par rapport au centre, tandis qu'une orbitale antiliante σ* est σu, car construite par différence d'orbitales atomiques s, ce qui est antisymétrique par rapport au centre.
Une orbitale liante de symétrie π est en revanche πu car l'inversion par rapport au centre de symétrie conduit à un changement de phase, tandis qu'une orbitale antiliante π* est πg car les deux orbitales atomiques p sont antisymétriques[7].
Liaisons chimiques et orbitales moléculaires
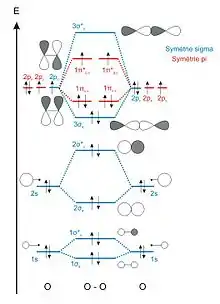
g : les orbitales πu,x et πu,y liantes et π*g,x et π*g,y antiliantes sont dégénérées.
Orbitales dégénérées
Des orbitales moléculaires sont dites dégénérées lorsqu'elles ont la même énergie. Ainsi, dans les molécules diatomiques homonucléaires des dix premiers éléments, les orbitales moléculaires dérivées des orbitales atomiques px et py donnent deux orbitales liantes dégénérées (de basse énergie) et deux orbitales antiliantes dégénérées (de haute énergie).
Liaison ionique
Lorsque la différence de niveaux d'énergie entre orbitales atomiques de deux atomes est suffisamment élevée — cas d'une forte différence d'électronégativité entre éléments — les orbitales de l'un des atomes contribuent presque entièrement aux orbitales liantes, tandis que les orbitales de l'autre atome contribuent presque entièrement aux orbitales antiliantes. Dans ce cas, un ou plusieurs électrons sont de facto transférés d'un atome à l'autre, ce qui donne une liaison ionique.
Ordre de liaison
L'ordre de liaison, ou indice de liaison, est le nombre de liaisons chimiques entre deux atomes. Il peut être établi à partir du nombre d'électrons impliqués dans les orbitales liantes et antiliantes. Un doublet d'électrons dans une orbitale liante ajoute une liaison, tandis qu'un doublet d'électrons dans une orbitale antiliante retranche une liaison. Par exemple, dans la molécule d'azote N2, de configuration électronique 1σ2
g 1σ2
u 2σ2
g 2σ2
u 1π4
u 3σ2
g, on compte huit électrons de valence dans des orbitales liantes (2σg, 1πu et 3σg) et deux électrons de valence dans une orbitale antiliante (2σu), d'où un ordre de liaison égal à trois, correspondant à une liaison triple.
L'énergie de liaison croît proportionnellement à l'ordre de liaison, tandis que la longueur de liaison évolue en sens inverse : une liaison chimique est d'autant plus courte qu'elle est forte.
L'ordre de liaison dans une molécule stable est supérieur à 0. Il existe cependant de rares molécules présentant une liaison chimique d'ordre nul. C'est notamment le cas de la molécule de dibéryllium Be2, qui présente une configuration électronique 1σ2
g 1σ2
u 2σ2
g 2σ2
u dans laquelle chaque orbitale liante σg est annulée par une orbitale antiliante σu ; cependant, les orbitales 2s et 2p peuvent s'hybrider, rendant l'orbitale 1σu légèrement moins antiliante, d'où une résultante 1σg + 1σu légèrement liante, de sorte que la molécule Be2, quoique très instable, a été observée en phase gazeuse, avec une longueur de liaison de 245 pm[12].
Orbitales frontières
Les orbitales frontières sont deux orbitales moléculaires particulières définies comme suit :
- l'orbitale haute occupée (HO, dite highest occupied molecular orbital, ou HOMO, en anglais) est l'orbitale moléculaire occupée de plus haute énergie ;
- l'orbitale basse vacante (BV, dite lowest unoccupied molecular orbital, ou LUMO, en anglais) est l'orbitale moléculaire vacante de plus basse énergie.
La différence d'énergie entre ces deux orbitales est appelée gap et peut être une mesure de l'excitabilité d'une molécule : plus ce gap est étroit, plus la molécule est excitable.
Exemples d'orbitales moléculaires
Dihydrogène H2
La molécule de dihydrogène H2 est l'exemple le plus simple de molécule diatomique homonucléaire, dont les deux atomes sont désignés H’ et H’’. Les orbitales atomiques de plus basse énergie, 1s’ et 1s’’, ne se transforment pas selon les symétries de la molécule. Toutefois, les orbitales atomiques adaptées à la symétrie peuvent former les combinaisons suivantes :
- 1s’ + 1s’’, combinaison symétrique : invariante par toutes les opérations de symétrie ;
- 1s’ – 1s’’, combinaison antisymétrique : signe inversé par réflexion, combinaison invariante par d'autres opérations de symétrie.
La combinaison symétrique, dite orbitale liante, a une énergie plus basse que les orbitales atomiques de base dont elle est issue, tandis que la combinaison antisymétrique, dite orbitale antiliante, a une énergie plus élevée que les orbitales de la base. La molécule H2 ayant deux électrons, ces derniers peuvent occuper l'orbitale symétrique σg et réduire l'énergie totale du système, qui devient ainsi plus stable que deux atomes d'hydrogène libres : c'est ce que l'on appelle une liaison covalente. En revanche, lorsque la molécule d'hydrogène est excitée, l'un des électrons de l'orbitale symétrique σg liante peut acquérir suffisamment d'énergie pour occuper l'orbitale antisymétrique σu antiliante, brisant la liaison covalente établie entre les deux atomes d'hydrogène, de sorte que la molécule H2 peut ainsi se dissocier.
Dihélium He2 et dibéryllium Be2
La molécule hypothétique de dihélium He2 diffère de celle de dihydrogène H2 par le fait qu'elle contiendrait quatre électrons, et non deux. Ces quatre électrons se répartiraient à raison de deux électrons sur l'orbitale symétrique σg liante et deux autres électrons sur l'orbitale antisymétrique σu antiliante, d'où une configuration électronique 1σ2
g 1σ2
u. La géométrie de la densité de probabilité de présence de ces électrons autour des deux noyaux d'hélium ne conduit pas la formation d'une liaison covalente, car si les électrons σg se localisent préférentiellement entre les noyaux atomiques, ce qui tend à les attirer l'un vers l'autre, les électrons σu se localisent quant à eux préférentiellement à l'opposé, c'est-à-dire derrière chaque noyau, à l'extérieur de la molécule, ce qui tend à écarter les noyaux l'un de l'autre. L'ordre de liaison rend compte de cet effet : il est nul dans le cas de la molécule He2, ce qui signifie que cette molécule n'existe pas.
Le cas de la molécule de dibéryllium Be2 est plus subtil. La même analyse que pour le dihélium conduit à la configuration électronique 1σ2
g 1σ2
u 2σ2
g 2σ2
u dans laquelle chaque orbitale liante σg est annulée par une orbitale antiliante σu, de sorte que, comme dans le cas du dihélium, l'ordre de liaison de cette molécule est nul, et la molécule ne devrait donc pas exister. Les orbitales 2s et 2p peuvent néanmoins s'hybrider, ce qui a pour effet de réduire le caractère antiliant de l'orbitale 1σu, d'où une résultante 1σg + 1σu légèrement liante : la molécule Be2, bien qu'elle soit très instable, a ainsi été observée en phase gazeuse, avec une longueur de liaison de 245 pm[12].
Dilithium Li2
Le cas de la molécule de dilithium Li2 est plus simple : la configuration électronique des atomes de lithium étant 1s2 2s1, celle de la molécule de dilithium est 1σ2
g 1σ2
u 2σ2
g. Avec quatre électrons sur des orbitales liantes (1σg et 2σg) et deux électrons sur une orbitale antiliante (1σu), l'ordre de liaison est ici de 1, ce qui correspond à une liaison simple
Les couches intérieures
La réactivité chimique repose principalement sur les électrons de la couche extérieure des atomes, appelés électrons de valence. Ainsi, les électrons décrits par les orbitales internes ne sont pas toujours pris en compte pour cette étude.
Une conséquence pratique est que si l'on exprime une orbitale moléculaire par une combinaison linéaire d'orbitales atomiques, les orbitales des couches intérieures peuvent souvent être omises sans influence majeure sur les résultats. Néanmoins, en vertu du principe variationnel, il s'agit d'une approximation : ajouter des orbitales dans la combinaison linéaire ne peut qu'améliorer la fonction d'onde (au pire, les coefficients des orbitales introduites seront nuls), ce qui conduira à une orbitale moléculaire de meilleure qualité (ce qui améliorera également a priori la fonction d'onde).
Approche plus quantitative
Pour obtenir des valeurs quantitatives pour les niveaux d'énergie moléculaires, on a besoin d'orbitales moléculaires telles que le développement de l'interaction de configuration converge rapidement vers la limite de l'interaction de configuration complète. La méthode la plus commune pour obtenir de telles fonctions est la méthode de Hartree-Fock qui exprime les orbitales moléculaires comme des fonctions propres de l'opérateur de Fock. On résout habituellement ce problème en développant les orbitales moléculaires comme une combinaison linéaire de fonctions gaussiennes centrées sur les noyaux atomiques (voir combinaison linéaire d'orbitales atomiques). L'équation pour les coefficients de ces combinaisons linéaires est une équation aux valeurs propres généralisée connue sous le nom de l'équation de Roothaan, qui est en fait une représentation particulière de l'équation de Hartree-Fock.
Notes et références
- (de) Friedrich Hund, « Zur Deutung einiger Erscheinungen in den Molekelspektren », Zeitschrift für Physik, vol. 36, nos 9-10, , p. 657-674 (DOI 10.1007/BF01400155, Bibcode 1926ZPhy...36..657H, lire en ligne)
- (de) Friedrich Hund, « Zur Deutung der Molekelspektren. I », Zeitschrift für Physik, vol. 40, no 10, , p. 742-764 (DOI 10.1007/BF01400234, Bibcode 1927ZPhy...40..742H, lire en ligne)
- (de) Friedrich Hund, « Zur Deutung der Molekelspektren V », Zeitschrift für Physik, vol. 63, nos 11-12, , p. 719-751 (DOI 10.1007/BF01339271, Bibcode 1930ZPhy...63..719H, lire en ligne)
- (en) Robert S. Mulliken, « Electronic States and Band Spectrum Structure in Diatomic Molecules. IV. Hund's Theory; Second Positive Nitrogen and Swan Bands; Alternating Intensities », Physical Review, vol. 29, no 5, , p. 637-649 (DOI 10.1103/PhysRev.29.637, Bibcode 1927PhRv...29..637M, lire en ligne)
- (en) Robert S. Mulliken, « The Assignment of Quantum Numbers for Electrons in Molecules. I », Physical Review, vol. 32, no 2, , p. 186-222 (DOI 10.1103/PhysRev.32.186, Bibcode 1928PhRv...32..186M, lire en ligne)
- (en) John Lennard-Jones, « The electronic structure of some diatomic molecules », Transactions of the Faraday Society, vol. 25, , p. 668-686 (DOI 10.1039/TF9292500668, lire en ligne)
- (en) Catherine E. Housecroft, Alan G. Sharpe, Inorganic Chemistry, Pearson Prentice Hall, 2e édition, 2005, p. 29-33.
- (en) Peter Atkins, Julio De Paula, Atkins’ Physical Chemistry, Oxford University Press, 8e édition, 2006.
- (en) Yves Jean, François Volatron, An Introduction to Molecular Orbitals, Oxford University Press, 1993.
- (en) Michael Munowitz, Principles of Chemistry, Norton & Company, 2000, p. 229-233.
- (en) Laura Gagliardi et Björn O. Roos, « Quantum chemical calculations show that the uranium molecule U2 has a quintuple bond », Nature, vol. 433, no 7028, , p. 848-851 (DOI 10.1038/nature03249, Bibcode 2005Natur.433..848G, lire en ligne)
- (en) V. E. Bondybey, « Electronic structure and bonding of Be2 », Chemical Physics Letters, vol. 109, no 5, , p. 436-441 (DOI 10.1016/0009-2614(84)80339-5, Bibcode 1984CPL...109..436B, lire en ligne)
Articles connexes
- Théorie de l'orbitale moléculaire
- Orbitale atomique
- Configuration électronique
- Orbitales frontières
- Chimie quantique
 Portail de la chimie
Portail de la chimie  Portail de la physique
Portail de la physique