Réarrangement de Pummerer
Le réarrangement de Pummerer est une réaction organique par laquelle un sulfoxyde d' alkyle se réarrange en un α- acyloxy - thioéther (mono thioacétal -ester) en présence d'anhydride acétique[1],[2],[3].
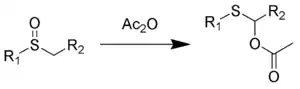
La stœchiométrie de la réaction est:
- RS(O)CHR'2 + Ac2O → RSC(OAc)R'2 + AcOH
Implémentation synthétique
L'anhydride acétique, l'anhydride trifluoroacétique et l'anhydride triflique ont été utilisés comme activateurs[6]. Outre les acétates, les nucléophiles les plus courants sont les arènes, les alcènes, les amides et les phénols.
L'utilisation de α-acylsulfoxydes et d'acide de Lewis, tels que TiCl4 et SnCl4, permet à la réaction de se dérouler à des températures plus basses (0 °C)[7].
Le chlorure de thionyle peut être utilisé à la place de l'anhydride acétique pour déclencher l'élimination pour former l'intermédiaire électrophile et fournir du chlorure comme nucléophile pour donner un α-chloro-thioéther[8] :
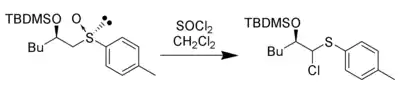
D'autres anhydrides et halogénures d'acyle peuvent donner des produits similaires. Les acides inorganiques peuvent également donner cette réaction. Ce produit peut être transformé en aldéhyde ou en cétone par hydrolyse[9].
Mécanisme
Le mécanisme du réarrangement de Pummerer commence par l'acylation du sulfoxyde (structures de résonance 1 et 2) par l'anhydride acétique pour donner 3, avec de l'acétate comme sous-produit. L'acétate agit alors comme catalyseur pour induire une réaction d'élimination pour produire la structure cationique-thial 4, avec de l'acide acétique comme sous-produit. Enfin, l'acétate attaque le thial pour donner le produit final 5.
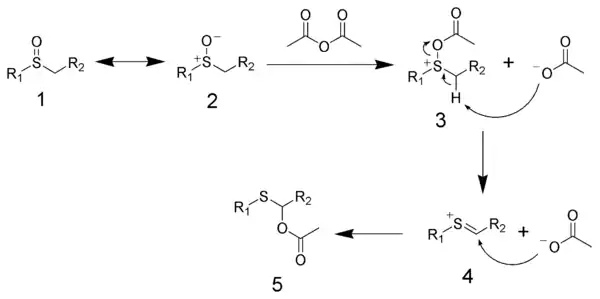
L'électrophile thial activé peut être piégé par divers nucléophiles intramoléculaires et intermoléculaires pour former des liaisons carbone-carbone et des liaisons carbone-hétéroatome.
L'intermédiaire est si électrophile que même les nucléophiles neutres peuvent être utilisés, y compris les cycles aromatiques avec des groupes donneurs d'électrons comme le 1,3-benzodioxole (en)[10] :
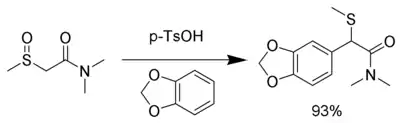
Il est possible d'effectuer le réarrangement en utilisant du sélénium à la place du soufre[11].
Fragmentation de Pummerer
Lorsqu'un substituant sur la position α peut former un carbocation stable, ce groupe plutôt que l'atome d'hydrogène α sera éliminé dans l'étape intermédiaire. Cette variante est appelée une « fragmentation Pummerer »[12]. Un exemple de ce type de réaction est la réaction entre un sulfoxyde et l'anhydride trifluoroacétique (TFAA):
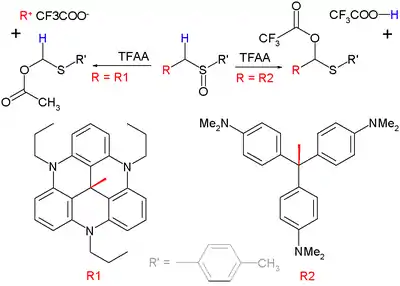
Le groupe organique R2 montré dans le diagramme ci-dessus en bas à droite est le carbocation du violet de méthyle, dont le pK R + de 9,4 n'est pas suffisant pour compenser la perte de H+ et donc un réarrangement classique de Pummerer se produit. La réaction de gauche est une fragmentation car le groupe partant avec pKR + = 23,7 est particulièrement stable.
Voir également
- Composé organosulfuré
- Réaction de Polonovski, réaction similaire impliquant un oxyde d'amine
Références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Pummerer rearrangement » (voir la liste des auteurs).
- Ottorino de Lucchi, Umberto Miotti et Giorgio Modena, The Pummerer Reaction of Sulfinyl Compounds, vol. 40, , 157–184 p. (ISBN 978-0471264187, DOI 10.1002/0471264180.or040.03)
- Padwa, Gunn et Osterhout, « Application of the Pummerer Reaction Toward the Synthesis of Complex Carbocycles and Heterocycles », Synthesis, vol. 1997, no 12, , p. 1353–1377 (DOI 10.1055/s-1997-1384)
- Padwa, Bur, Danca et Ginn, « Linked Pummerer-Mannich Ion Cyclizations for Heterocyclic Chemistry », Synlett, vol. 2002, no 6, , p. 851–862 (DOI 10.1055/s-2002-31891)
- Pummerer, « Über Phenyl-sulfoxyessigsäure », Chemische Berichte, vol. 42, no 2, , p. 2282–2291 (DOI 10.1002/cber.190904202126)
- Pummerer, « Über Phenylsulfoxy-essigsäure. (II.) », Chemische Berichte, vol. 43, no 2, , p. 1401–1412 (DOI 10.1002/cber.19100430241, lire en ligne)
- Smith, Coote, Sneddon et Procter, « Beyond the Pummerer Reaction: Recent Developments in Thionium Ion Chemistry », Angewandte Chemie International Edition, vol. 49, no 34, , p. 5832–44 (PMID 20583014, DOI 10.1002/anie.201000517).
- Stamos, « Arylation of α-phosphoryl sulfides via their pummerer rearrangement intermediates generated from the corresponding sulfoxides », Tetrahedron Letters, vol. 27, no 51, , p. 6261–6262 (DOI 10.1016/S0040-4039(00)85447-7).
- Kosugi, Watanabe et Uda, « Lewis Acid-Mediated Carbon-Carbon bond forming reaction using the Pummerer Rearrangement Products from Chiral beta-Hydroxy Sulfoxides », Chemistry Letters, vol. 18, no 10, , p. 1865–1868 (DOI 10.1246/cl.1989.1865)
- Meffre, Durand et Le Goffic, « Methyl (S)-2-phthalimido-4-methylthiobutanoate », Organic Syntheses, vol. 76, , p. 123 (DOI 10.15227/orgsyn.076.0123).
- Ishibashi, Miki, Ikeda et Kiriyama, « Synthesis of α-(Methylthio)arylacetamides and Their Conversion into Some Biologically Active Arylethylamines », Biological & Pharmaceutical Bulletin, vol. 37, no 12, , p. 3396–3398 (DOI 10.1248/cpb.37.3396).
- Gilmour, Prior, Burton et Holmes, « An organocatalytic approach to the core of eunicellin », Chemical Communications, no 38, , p. 3954 (DOI 10.1039/B709322E).
- Laleu, Santarém Machado et Lacour, « Pummerer fragmentation vs. Pummerer rearrangement: a mechanistic analysis », Chemical Communications, no 26, , p. 2786–2788 (DOI 10.1039/b605187a)
 Portail de la chimie
Portail de la chimie