Structure de l'ARN
La structure de l'ARN décrit l'arrangement des paires de bases et de la conformation de l'ARN en trois dimensions. L'ARN étant trouvé le plus souvent sous forme de simple-brin dans la cellule, il se replie en effet sur lui-même en formant des appariements Watson-Crick intramoléculaires. Ceci conduit à la formation de régions localement en hélice et de régions en boucle où les bases ne sont pas appariées. Cette topologie des appariements constitue ce qu'on appelle la structure secondaire de l'ARN[2]. En plus de ces appariements standard, l'ARN peut former des interactions non canoniques et des interactions à longue distance qui contribuent à donner un repliement 3D à certains ARN structurés, comme les ARNt ou les ARN ribosomiques, on parle alors de la structure tertiaire des ARN.
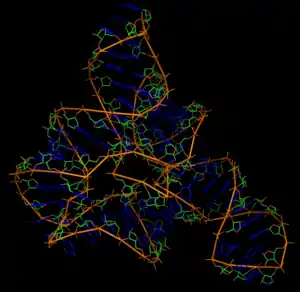
L'existence de structures secondaires et tertiaires bien définies dans les ARN est un des éléments importants de la fonction d'un certain nombre d'entre eux. Ces structures leur permettent de former des sites de liaison pour des ligands sélectifs, petites molécules ou protéines, et, pour les ribozymes, elles leur permettent d'assurer des fonctions catalytiques. La formation ou la fusion de ces structures en réponse à une variation de l'environnement peut-être aussi être un signal déclenchant une réponse cellulaire.
L'analyse et la prédiction de la structure des ARN, et en particulier de leur structure secondaire, est un champ de recherche très actif, à la fois dans le domaine de la biologie moléculaire et de la bio-informatique. En particulier, l'existence de règles plus formalisées que pour la structure des protéines (dans les paires Watson-Crick, A s'apparie avec U, et G avec C) est une des raisons du succès de ces méthodes prédictives.
Structure secondaire
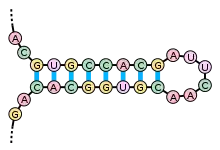
La structure secondaire d'un ARN est la description de l'ensemble des appariements internes au sein d'une molécule simple brin. Cet ensemble d'appariements induit une topologie particulière, composée de régions en hélice (tiges) et de régions non appariées (boucles). Par extension, la structure secondaire recouvre également la description de cette topologie.
L'élément moteur de la formation de structures secondaires au sein d'un ARN simple-brin est l'existence de régions contenant des séquences répétées inversées, qui peuvent s'apparier pour former localement une structure en double hélice. Par exemple, si l'ARN contient les deux séquences suivantes : --GUGCCACG----CGUGGCAC--, celles-ci forment un motif répété inversé, les nucléotides du second segment étant les complémentaires de ceux du premier, après inversion de leur sens de lecture. Ces deux segments peuvent donc s'apparier de manière antiparallèle pour former une région localement en duplex. La région entre les deux segments forme alors une boucle reliant les deux brins du duplex. On parle alors de structure en tige et boucle ou en épingle à cheveux.
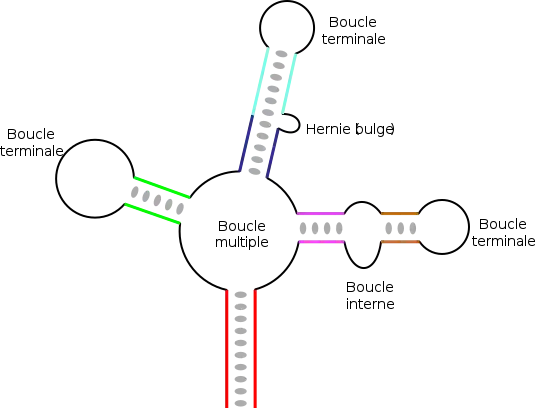
Dans des ARN de longueur plus importante, il peut exister des structures plus complexes, qui résultent de l'appariement de plusieurs régions complémentaires ou séquences répétées inversées. En fonction de la manière dont sont « emboîtées » ces différentes régions, on obtient des éléments topologiques variés, avec des tiges ou régions appariées, et divers types de boucles :
- les boucles terminales, situées à l'extrémité d'une tige ;
- les boucles internes, qui connectent deux tiges ;
- les boucles multiples, qui connectent trois tiges ou plus et constituent des points de branchement de la structure ;
- les hernies (en anglais bulge) ou boucles latérales qui sont sur un seul des deux brins d'une hélice. La continuité de l'hélice n'est en général pas affectée et l'ensemble des bases reste empilé de manière coaxiale, de part et d'autre de la hernie.
Il n'existe pas toujours une structure unique stable pour une séquence donnée et il arrive que certains ARN puissent adopter plusieurs conformations alternatives en fonction de la liaison d'un ligand (protéine, petite molécule…) ou des conditions physico-chimiques (force ionique, pH).
Déterminants de stabilité
La stabilité de la structure des ARN est principalement déterminée par trois types d'interactions :
- une contribution électrostatique liée à la formation des liaisons hydrogène entre les bases ;
- l'empilement des plateaux des paires de bases (stacking) qui donne naissance à des interactions de van der Waals ;
- l'interaction avec le solvant aqueux et les ions de la solution. En particulier, la présence de cations divalents comme le magnésium, Mg2+, est souvent nécessaire pour neutraliser la charge des groupements phosphate du squelette de l'ARN. Le repliement et la formation d'une structure tridimensionnelle par l'ARN implique en particulier le rapprochement de ces groupements phosphate, ce qui serait électrostatiquement défavorable en l'absence de contre-ions.
Le groupement 2'-hydroxyle (-OH) porté spécifiquement par les riboses de l'ARN (mais absent dans l'ADN) joue parfois également un rôle dans la stabilité et la formation de la structure, en formant des liaisons hydrogène avec les bases.
Paramètres thermodynamiques
L'étude de la stabilité des ARN est basée sur la mesure de courbes de fusion par spectrophotométrie, en suivant l'effet hyperchrome[3] dont on peut extraire les paramètres thermodynamiques de la formation des paires de bases. On utilise en général des courts oligonucléotides de 10 à 20 bases de long, ce qui permet d'avoir des températures de fusion comprises entre 30 et 70 degrés Celsius.
Dans l'exemple simplifié d'un oligonucléotide de séquence palindromique, qui est donc son propre brin complémentaire, cela revient à étudier l'équilibre :

La résolution des équations d'équilibre permet de calculer la température de fusion, en fonction des valeurs standards des fonctions d'état (enthalpie et entropie). On trouve :


Avec R, la constante des gaz parfaits et , la concentration totale de brins d'ARN (appariés ou non) dans la solution.

En mesurant expérimentalement la température de fusion pour plusieurs valeurs de concentration de l'oligonucléotide, il est possible, par ajustement de l'expression ci-dessus, de déterminer les valeurs de l'entropie et de l'enthalpie standard ( et ) et donc de l'enthalpie libre à la température souhaitée.



En mesurant cette valeur pour un ensemble suffisamment important et diversifié de courts duplex d'ARN, il est possible de définir des règles permettant de calculer a priori l'enthalpie libre standard associée à la formation de chaque paire de base. Dans la pratique, pour tenir compte des interactions d'empilement entre bases consécutives, on estime empiriquement la variation d'entalphie libre standard () obtenue lorsqu'on empile une paire de base X-Y en 3' d'une paire de base V-W. Cette variation d'enthalpie libre combine la contribution liée à la formation des liaisons hydrogène entre les deux bases de l'appariement additionnel et la contribution d'empilement, qui dépend de la nature des paires X-Y et V-W.

Ces paramètres ont été tabulés pour différentes conditions de tampon[4],[5] et peuvent être utilisées pour calculer l'enthalpie libre standard associée à la formation d'une hélice d'ARN, avec une marge d'incertitude de l'ordre de 10 %. La table suivante donne les valeurs obtenues à 37 °C, en présence d'une concentration de 1M NaCl (règles de Freier-Turner[5]). À ces valeurs, il faut ajouter un coût de nucléation, d'origine entropique, de +3,4 kcal/mol.
| séquence | 5'AA3'
3'UU5' |
5'AU3'
3'UA5' |
5'UA3'
3'AU5' | 5'CA3'
3'GU5' |
5'CU3'
3'GA5' |
5'GA3'
3'CU5' |
5'GU3'
3'CA5' |
5'CG3'
3'GC5' |
5'GC3'
3'CG5' |
5'GG3'
3'CC5' |
|---|---|---|---|---|---|---|---|---|---|---|
| kcal/mol | -0,9 | -0,9 | -1,1 | -1,8 | -1,7 | -2,3 | -2,1 | -2,0 | -3,4 | -2,9 |
On peut ainsi calculer a priori l'enthalpie libre standard associé à la formation d'une hélice d'ARN, en additionnant tous les d'empilement. Par exemple, pour le duplex de 7 paires de bases suivant, il faut additionner les 6 d'empilement et le coût de nucléation.
5'-AGGCUUC-3' 3'-UCGCAAG-5'
En prenant les valeurs de la table ci-dessus, on trouve = - 1,7 - 2,9 - 3,4 - 1,7 - 0,9 - 2,3 + 3,4nuc = -9,5 kcal/mol
Des règles ont également été déterminées pour les différents types de boucles, en fonction de leur longueur. Ces calculs d'enthalpie libre sur les boucles sont toutefois d'une précision moindre, en particulier parce que certaines boucles peuvent adopter des conformations stables particulières[6] qui ne sont pas prises en compte dans ce modèle simplifié. Cet ensemble de règles empiriques est exhaustif, il permet faire un calcul prédictif de l'enthalpie libre associée à n'importe quelle structure secondaire.
Analyse et prédiction de structure secondaire
Un ARN de longueur suffisante peut en principe adopter un grand nombre de conformations différentes, correspondant à un grand nombre de structures secondaires alternatives différentes. En théorie, la configuration la plus stable est celle qui correspond à l'enthalpie libre standard minimale. Les calculs thermodynamiques décrits ci-dessus permettent de calculer théoriquement cette enthalpie, pour toute configuration de structure secondaire d'un ARN donné. Le problème qui est posé est trouver la structure d'énergie minimale parmi toutes les configurations possibles. Le nombre de combinaisons possibles est cependant très grand, pour une structure de longueur 2 n, il croit comme le nombre de Catalan :

De plus, l'imprécision sur les calculs empiriques d'énergie rend la prédiction difficile. Pour résoudre ce problème, on associe en général des méthodes expérimentales, comparatives et bio-informatiques. Les approches combinées permettent ainsi d'obtenir des analyses fiables, où la prédiction est corroborée par des arguments expérimentaux et évolutifs.
Méthodes expérimentales
Il est possible d'analyser la structure secondaire des ARN au moyen de méthodes chimiques et enzymatiques. Cette technique est basée sur la réactivité différentielle de l'ARN, en fonction de son état de structuration. Par exemple, certaines ribonucléases, comme la nucléase S1, ne clivent l'ARN que dans les régions qui sont simple brin, tandis que d'autres, à l'inverse, ne coupent que dans les régions en double-brin. On traite l'ARN étudié par l'une ou l'autre de ces enzymes dans des conditions de digestion très partielle, où il ne se produit que zéro ou une coupure par molécule d'ARN. En analysant par électrophorèse les fragments d'ARN ainsi produits, on peut localiser les sites de coupure et donc les régions qui sont en duplex ou bien non appariées.
L'utilisation de sondes chimiques permet d'avoir une information encore plus précise, en localisant quelles positions de chaque base sont accessibles au réactif. On utilise par exemple du sulfate de diméthyle (DMS) qui réagit avec la position N3 de la cytidine et la position N7 de la guanine. La première, le N3 dans les C, est impliquée dans une liaison hydrogène avec le G dans des appariements Watson-Crick et n'est réactive que si le C correspondant n'est pas apparié[7].
Méthodes phylogénétiques

Lorsqu'on compare les ARN remplissant la même fonction chez plusieurs espèces, on observe en général une conservation forte de la structure secondaire. Les différentes régions en hélice et leur topologie est conservée même si la nature exacte des appariements de bases qui les constituent peut varier. Par exemple, à une position donnée, on pourra trouver un appariement G-C dans une espèce, remplacé par un appariement A-U dans une autre, ce qui ne modifie pas la topologie globale. Cette contrainte d'appariement est une conséquence de la pression de sélection sur la fonction de l'ARN, qui est en général fortement dépendante de sa structure tridimensionnelle : pour conserver la fonction, il faut conserver la forme et donc la topologie.
Dans des alignements de séquence d'ARN homologues, cette conservation structurale globale de la conduit à l'apparition de covariations entre positions qui sont normalement appariées dans le repliement. Ces covariations peuvent être identifiées systématiquement par des analyses statistiques sur un ensemble de séquences d'ARN homologues et utilisées pour construire des modèles de structure secondaire. Il est également possible de vérifier a posteriori si un modèle de structure secondaire est compatible avec les covariations observées dans l'évolution. C'est en grande partie ainsi qu'ont été validés les modèles de structure secondaire des ARN ribosomiques[8].
Prédictions bio-informatiques
La disponibilité de données expérimentales permettant de calculer l'enthalpie libre associée à un repliement donné de l'ARN a ouvert la voie à la prédiction ab initio de sa structure secondaire. L'objectif étant de trouver la ou les conformations d'énergie minimale, qui correspondent à l'état le plus stable du système.
Un des premiers algorithmes performants pour prédire la structure secondaire de l'ARN a été développé par Ruth Nussinov[9] et est basé sur l'utilisation de la programmation dynamique pour limiter la complexité combinatoire. L'algorithme de Nussinov ne cherche qu'à maximiser le nombre total d'appariements de base dans la structure formée, sans critères énergétiques, ni prise en compte des interactions d'empilements entre paires de bases. Une version améliorée de cet algorithme de base a été proposée en 1981 par Michael Zuker, qui incorpore les données thermodynamiques et en particulier les interactions d'empilement[10].
Ces deux algorithmes, très proches dans leur principe, sont performants et permettent de prédire le repliement optimal d'un ARN jusqu'à quelques milliers de nucléotides de longueur. Leur complexité algorithmique est de l'ordre de O(N3), où N est la longueur de la séquence. Tous deux souffrent cependant de deux limitations importantes :
- ils ne permettent de prédire que des topologies canoniques, excluant en particulier la possibilité de former des pseudonœuds. Ceci n'est pas un défaut majeur, car les pseudonœuds sont à la fois peu fréquents et de longueur limitée (voir plus bas). On peut donc en général les ajouter a posteriori sur la prédiction de structure classique de type Zuker/Nussinov ;
- ils ne prédisent que la structure d'énergie théorique minimale, et pas les solutions dont le score énergétique peut être très proche de l'optimum. C'est une limitation importante, car les paramètres énergétiques empiriques ne sont précis qu'au mieux à 5 à 10 % près, et, à cause de cette incertitude, la structure la plus stable en réalité peut se trouver dans ces solutions légèrement sous-optimales.
Un algorithme permettant la prédiction de structures sous-optimales a ensuite été développé par Zuker[11] et constitue la version actuelle du programme mfold, disponible en ligne, qui est actuellement un des outils standards de la prédiction de structure secondaire d'ARN.
Plusieurs variantes de ce programme existent, et permettent d'affiner les prédictions, en tenant compte de données phylogénétiques ou expérimentales, ainsi que de la reconnaissance de motifs structuraux spécifiques, comme les tétraboucles (voir ci-après).
Structure tertiaire
En plus de leur structure secondaire, constituée d'appariements de base standard, les ARN peuvent adopter une structure tridimensionnelle compacte, bien définie, qui résulte d'interactions additionnelles entre les éléments classiques de structure secondaire (hélices, boucles). Ces interactions additionnelles dans la plupart des cas de type non canonique, c'est-à-dire qu'il ne s'agit ni d'appariement Watson-Crick, ni d'appariements bancals, de type G-U.
Les appariements non canoniques et les interactions additionnelles à longues distances sont relativement bien connus, en particulier grâce à la résolution de la structure tridimensionnelle du ribosome, qui en a mis en lumière un très grand nombre. L'utilisation de méthodes phylogénétiques (voir ci-dessus) permet également de repérer certaines de ces interactions non canoniques, lorsqu'on dispose de la séquence du même ARN chez plusieurs espèces. Il est ainsi possible de rationaliser en partie la manière dont se construit la structure tertiaire des ARN et de réaliser des prédictions de repliement relativement fiables, dont certaines ont pu être validées a posteriori par l'expérience.
Appariements non canoniques
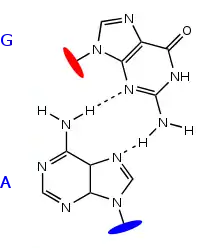
Les appariements non canoniques sont des interactions entre nucléotides, distincts des d'appariements de base classiques, de type Watson-Crick (A-U et G-C) ou bancals (wobble, G-U). On a observé une grande variété de ces appariements dans les structures tridimensionnelles d'ARN résolues par cristallographie ou par RMN. Ces appariements non canoniques impliquent toujours des liaisons hydrogène entre les bases, qui sont coplanaires, comme dans les paires Watson-Crick.
Une nomenclature systématique de toutes ces interactions a été proposée par Eric Westhof et ses collaborateurs[12]. Plus de 150 types d'appariements ont été observés et ont été regroupés en douze grandes familles.

Cette classification repose entre autres sur la face des bases impliquée dans l'interaction. Un nucléotide de type purine comporte par exemple trois faces permettant des liaisons hydrogène :
- la face Watson-Crick. C'est le côté comportant le N1 et les fonctions oxo et amino en position 2 et 6. C'est cette face qui est impliquée dans les appariements canoniques A-U et G-C, ainsi que dans les appariements bancals G-U ;
- la face Hoogsteen. C'est le côté comprenant le N7 et la fonction oxo ou amino en position 6 ;
- la face ribose. C'est le côté comprenant le N3 et la fonction en position 2. La fonction 2' hydroxyle du ribose peut également participer aux interactions sur cette face.
À cette nomenclature d'interactions entre bases, il faut ajouter la possibilité pour ces dernières d'adopter différentes conformations par rapport au ribose auquel elles sont liées. La liaison glycosidique qui relie la base au sucre a en effet deux orientations favorables principales, suivant que la face Watson-Crick de la base est située du côté du ribose (orientation cis) ou du côté opposé (orientation trans). Dans les appariements canoniques, les nucléotides sont toujours en conformation trans. Cette orientation du sucre par rapport à la base détermine en particulier la polarité, parallèle ou antiparallèle, des brins d'ARN qui sont appariés.
Parmi ces appariements non canoniques relativement fréquents, on trouve par exemple des appariements Hoogsteen A-U impliquant la face Hoogsteen d'une adénosine et un uracile, des paires G-A "en cisaille" (sheared), impliquant la face Hoogsteen du A et la face ribose du G[13].
Enfin, on trouve plus rarement des appariements qui impliquent des formes protonées de certaines bases. Ainsi, existe des paires C-C+, où la deuxième cytosine est protonée sur sa position N3. Ce type de paires de bases non canoniques porte une charge positive et est stabilisé à pH acide (le pKa du N3 de la cytidine est d'environ 4.2).
Motifs récurrents
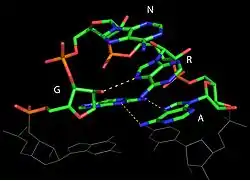
Tétraboucles
L'analyse de la structure des ARN ribosomiques a permis de mettre en évidence la présence d'un nombre très élevé de structures en hélices fermées par des boucles terminales de quatre nucléotides[15]. Ces boucles, appelées tétraboucles (tetraloop), ont des structures très stables, renforcées par des interactions non canoniques. On en a identifié plusieurs classes, dont les principales se regroupent dans trois familles :
- les boucles de type GNRA, ou N peut être A, G, C ou U, et R, A ou G. Dans ces boucles (figure ci-contre). Le G en 5' et le A en 3' forment une paire G-A en cisaille. La purine en position 3 est empilée sur l'adénine et fait une liaison avec le 2'-OH du ribose de l'adénine[13] ;
- les boucles de type UNCG, ou N peut être A, G, C ou U ;
- les boucles de type CUUG.
Les tétraboucles de la famille GNRA, qui sont parmi les motifs les plus fréquents dans l'ARN, peuvent former des interactions à longue distance avec des régions en hélice ou en boucle interne. Ces interactions impliquent les deux nucléotides au centre de la boucle qui sont exposés. Ainsi, par exemple, les tétraboucles de séquence GAAA peuvent interagir spécifiquement dans le petit sillon avec des hélices contenant deux paires G-C consécutives et en particulier avec le motif de 11 nucléotides [CCUAAG...UAUGG] où les deux premiers C sont appariés avec deux derniers G[16]. On appelle ces séquences des récepteurs de tétraboucle. Ces interactions tétraboucle-récepteur sont très fréquentes dans les structures tridimensionnelles d'ARN et jouent un rôle important dans le maintien du repliement global.
Boucle E
La boucle E est un motif qui a été identifié pour la première fois dans l'ARN ribosomique 5S. Sur les prédictions informatiques de structure secondaire, cette région avait été initialement postulée comme formant une boucle interne, encadrée par deux tiges appariées. Cette « boucle » étant résistante aux nucléases, on a ensuite conclu à la formation de structure non canonique, qui aboutissent à la formation d'une tige compacte. Elle est toujours appelée boucle E, bien qu'il ne s'agisse in fine pas du tout d'une structure en boucle.
La boucle E contient une paire A-A parallèle et un triplet de paire de bases, encadrés par une ou deux paires G-A en cisaille[17]. Ce motif a ensuite été retrouvé dans d'autres régions des ARN ribosomiques et dans d'autres ARN structurés, comme les viroïdes. Il existe des protéines qui reconnaissent sélectivement les motifs boucle E.
Pseudonœuds
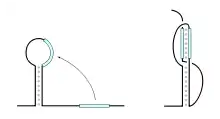
Un pseudonœud est une structure formée par l'interaction d'une boucle avec une région située à l'extérieur de la tige qui la délimite[18]. Les structures secondaires canoniques, décrites ci-dessus excluent en principe la formation de pseudonœuds, en particulier parce que leur prédiction par des méthodes bio-informatiques reste assez complexe.
Les pseudonœuds permettent de former des interactions à longue distance, entre deux régions non appariées dans la structure secondaire. Ce sont donc des éléments importants de la structure tridimensionnelle des ARN. Par rapport aux structures secondaires canoniques, ils sont en général peu nombreux et de dimension limitée dans les ARN structurés, mais ils jouent souvent un rôle important dans la structure et la fonction. La limite sur leur taille a une origine topologique, liée à la formation de l'hélice additionnelle du pseudonœud : celle-ci ne peut en principe pas dépasser un tour d'hélice, soit une dizaine de paires de bases, sinon cela conduirait à un enlacement complet des brins créant un nœud véritable.
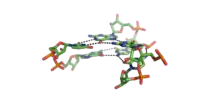
Interactions triples
Une interaction triple, ou triplet de bases, est une interaction entre trois bases situées dans le même plan et formant des liaisons hydrogène entre elles. Elles peuvent être isolées ou se produire de manière consécutive, les triplets formant alors des empilements, comme les paires de bases dans un duplex. On a alors une structure associant localement trois brins d'ARN.
Le type le plus fréquent d'interactions triples s'observe au sein d'une hélice classique, lorsque l'un des deux brins contient plusieurs purines consécutives. Un troisième brin d'ADN peut alors former des appariements Hoogsteen avec le brin purique, en s'insérant dans le grand sillon de l'hélice.
Le cœur classique de la structure des ARN de transfert contient plusieurs interactions triples qui stabilisent l'interaction des quatre hélices et donnent à ces ARN leur topologie globale en forme de « L » à partir de leur structure secondaire en forme de feuille de trèfle.
Notes et références
- (en) Montange R.K., Batey R.T., « Structure of the S-adenosylmethionine riboswitch regulatory mRNA element », Nature, vol. 441, no 7097, , p. 1172-1175 (PMID 16810258)
- (en) Doty P., Boedtker H., Fresco J. R. , Haselkorn R., Litt M., « Secondary Structure in Ribonucleic Acids », Proc. Natl. Acad. Sci. USA, vol. 45, no 4, , p. 482-499
- (en) Mergny J.L., Lacroix L., « Analysis of thermal metling curves », Oligonucleotides, vol. 13, no 6, , p. 515-537 (PMID 15025917)
- (en) Tinoco I. Jr., Borer P.N., Dengler B., Levin M.D., Uhlenbeck O.C., Crothers D.M., Bralla J., « Improved estimation of secondary structure in ribonucleic acids. », Nat. New Biol., vol. 246, no 150, , p. 40-41 (PMID 4519026)
- (en) Freier S.M., Kierzek R., Jaeger J.A., Sugimoto N., Caruthers M.H., Neilson T., Turner D.H., « Improved free-energy parameters for predictions of RNA duplex stability. », Proc. Natl. Acad. Sci. USA, vol. 83, no 24, , p. 9373-9377 (PMID 2432595)
- (en) Tuerk C., Gauss P., Thermes C., Groebe D.R., Gayle M., Guild N., Stormo G., d'Aubenton-Carafa Y., Uhlenbeck O.C., Tinoco I. Jr., « CUUCGG hairpins: extraordinarily stable RNA secondary structures associated with various biochemical processes. », Proc. Natl. Acad. Sci. USA, vol. 85, no 5, , p. 1364-1368 (PMID 2449689)
- (en) Peattie D.A., Gilbert W., « Chemical probes for higher-order structure in RNA. », Proc. Natl. Acad. Sci. USA, vol. 77, no 8, , p. 4679-4682 (PMID 6159633)
- (en) Noller H.F., Woese C.R., « Secondary structure of 16S ribosomal RNA. », Science, vol. 212, no 4493, , p. 403-411 (PMID 6163215)
- (en) Nussinov R., Pieczenik G., Griggs J. R., Kleitman D. J., « Algorithms for loop matchings. », SIAM J. Appl. Math., vol. 31, no 1, , p. 68-82
- (en) Zuker M., Stiegler P., « Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. », Nucleic acids res., vol. 9, no 1, , p. 133-148 (PMID 6163133)
- (en) Zuker M., « On finding all suboptimal foldings of an RNA molecule. », Science, vol. 244, no 4900, , p. 48-52 (PMID 2468181)
- (en) Leontis N.B., Stombaugh J., Westhof E., « The non-Watson-Crick base pairs and their associated isostericity matrices. », Nucleic Acids Res., vol. 30, no 16, , p. 3497-3531 (PMID 12177293)
- (en) Heus H.A., Pardi A., « Structural features that give rise to the unusual stability of RNA hairpins containing GNRA loops. », Science, vol. 253, no 5016, , p. 191-194 (PMID 1712983)
- (en) Cate, J.H., Gooding, A.R., Podell, E., Zhou, K., Golden, B.L., Kundrot, C.E., Cech, T.R., Doudna, J.A., « Crystal structure of a group I ribozyme domain: principles of RNA packing. », Science, vol. 273, , p. 1676–1685 (PMID 8781224)
- (en) Woese, C.R., Winkers, S., Gutell, R.R., « Architecture of ribosomal RNA: Constraints on the sequence of "tetra-loops" », Proc. Nati. Acad. Sci. USA, vol. 87, , p. 8467–71 (PMID 2236056, DOI 10.1073/pnas.87.21.8467)
- (en) Costa, M., Michel, F., « Frequent use of the same tertiary motif by self-folding RNAs. », EMBO J., vol. 14, , p. 1276–1285 (PMID 7720718)
- (en) Neocles B. Leontis et Eric Westhof, « A common motif organizes the structure of multi-helix loops in 16 S and 23 S ribosomal RNAs », J. Mol. Biol., vol. 283, , p. 571-583 (PMID 9784367)
- (en) D.W. Staple et S.E. Butcher, « Pseudoknots: RNA Structures with Diverse Functions. », PloS Biol., vol. 3, , e213 (PMID 15941360)
Voir aussi
Articles connexes
Lien externe
 Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire