Spectroscopie RMN
La spectroscopie RMN est une technique qui exploite les propriétés magnétiques de certains noyaux atomiques. Elle est basée sur le phénomène de résonance magnétique nucléaire (RMN), utilisé également en imagerie médicale sous le nom d’IRM.
Pour les articles homonymes, voir RMN.

Les applications les plus importantes pour la chimie organique sont la RMN du proton et du carbone 13 effectuée sur des solutions liquides. Mais la RMN est aussi applicable à tout noyau possédant un spin non nul, que ce soit dans les solutions liquides ou dans les solides. Certains gaz comme le xénon peuvent aussi être mesurés lorsqu'ils sont absorbés dans des matériaux poreux par exemple.
Contrairement à la spectroscopie RMN des solutions qui est utilisée de manière routinière dans les laboratoires académiques ou industriels, la RMN des solides reste légèrement moins abordable sans une connaissance plus approfondie du phénomène RMN.
Historique
La spectroscopie RMN naît en 1946 lorsque Felix Bloch et Edward Mills Purcell, de manière indépendante, réalisent les premières mesures du magnétisme nucléaire par induction magnétique. Ils reçoivent pour cette invention le prix Nobel de physique en 1952. Les développements sont ensuite conséquents : en 1950, Erwin L. Hahn découvre les échos de spin à la base des nombreuses techniques multi-impulsionnelles utilisées de nos jours[1]. La même année, W. Proctor et W. Dickinson découvrent indépendamment le phénomène de déplacement chimique, découverte fondamentale pour l'essor des applications de la RMN en chimie organique[2],[3]. En 1959, E.R. Andrew démontre que la rotation d'un échantillon autour d'un axe particulier, l'angle magique, permet l'obtention de spectres résolus en RMN des solides (RMN MAS)[4]. Enfin, une étape majeure dans le développement de la mesure du phénomène RMN est la conception de la spectroscopie RMN par transformée de Fourier par Richard R. Ernst en 1966. Il reçoit le prix Nobel de chimie en 1991 pour cette découverte et les développements de la RMN multidimensionnelle que cette technique a permis.
Principes de base
La spectroscopie RMN repose sur la détection du phénomène RMN, qui se produit lorsque des noyaux atomiques de spin non nuls sont placés dans un champ magnétique externe généralement uniforme et qu'ils sont excités par un rayonnement radiofréquence accordé sur les différences d'énergie entre les différents états possibles du spin nucléaire.
La fréquence de résonance (appelée fréquence de Larmor) est en première approximation directement proportionnelle au champ appliqué :



où est le rapport gyromagnétique.

Le fait que chaque isotope possède un rapport gyromagnétique unique permet à la technique RMN de pouvoir être réglée sur un élément particulier. Il suffit d'ajuster la fréquence d'excitation et d'observation sur le noyau ciblé.
La fréquence de résonance des noyaux dépend aussi de leur environnement, les spins étant en interaction avec celui-ci. Ces interactions sont appelées interactions internes par opposition aux interactions externes des spins avec le champ magnétique externe et le rayonnement radiofréquence. Ces interactions intra- ou intermoléculaires peuvent être magnétiques comme c'est le cas pour le déplacement chimique et les couplages dipolaires, ou électriques, ce qui est le cas de l'interaction quadripolaire. L'interprétation et la mesure de ces interactions permettent d'avoir des informations précieuses sur :
- la nature et le nombre d'atomes voisins des noyaux étudiés ;
- la liaison chimique ;
- la conformation moléculaire ;
- les distances interatomiques ;
- la mobilité moléculaire ;
- la configuration relative ou absolue de certains centres chiraux ;
- etc.
Origine du déplacement chimique
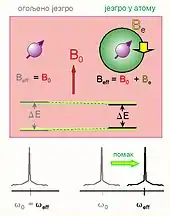
Quand une molécule est placée dans le champ magnétique externe , des champs magnétiques locaux sont créés par la circulation des électrons induite dans les diverses orbitales moléculaires sous l'action du champ. En effet une particule chargée se déplaçant dans un champ magnétique crée autour d'elle un champ magnétique local qui s'oppose, le plus souvent, au champ magnétique global (B0). Le champ réellement « ressenti » localement par les noyaux n'est donc plus exactement le champ appliqué, mais la somme de B0 et du champ magnétique créé par les électrons.
D'autres effets peuvent intervenir qui sont également susceptibles de modifier le champ local, comme le paramagnétisme électronique, lorsqu’il y a des électrons délocalisés (noyaux aromatiques, liaisons chimiques fortement polarisées, électrons non appariés…) ou non appariés qui vont au contraire augmenter le champ ressenti par les noyaux. C'est alors un effet paramagnétique.
La façon dont la fréquence de résonance de chaque noyau est affectée par ces champs locaux est caractérisée par une constante d'écran, , qui contient les contributions dia- et paramagnétiques :


La différence de fréquence δ induite par les différents environnements électroniques est généralement appelée déplacement chimique.
Les utilisateurs de la RMN parlent souvent de blindage et de déblindage (en anglais, shielding et deshielding ; shield signifiant « bouclier ») par référence à l’amplitude de la constante d’écran . Plus un noyau est entouré d'électrons, moins il ressent le champ magnétique global B0 et donc plus il est blindé et se retrouve sur la droite du spectre RMN (δ faible)[note 1]. À l'opposé, si un noyau est statistiquement appauvri en électrons par la présence d'un atome plus électronégatif ou d'un groupe mésomériquement attracteur, il est alors déblindé et se déplace vers la gauche du spectre (δ plus élevé). Ce concept est cependant peu recommandé car il néglige les effets paramagnétiques.
Les échelles de déplacement chimique
Il a toujours été primordial d'exprimer les déplacements chimiques indépendamment de l'intensité du champ magnétique utilisé surtout que les premiers spectromètres à « onde continue » utilisaient une fréquence d'observation constante (30, 60, 100 MHz, etc.) et un champ magnétique externe variable (aujourd'hui, avec les méthodes impulsionnelles, le champ magnétique est généralement fixe).
On a donc rapidement décidé de créer une échelle sans unité selon la formule suivante :
- (en ppm)

c'est-à-dire :

Comme la différence entre la fréquence de résonance et la fréquence de résonance de la référence est de l'ordre du hertz alors que la fréquence du spectromètre est de l'ordre du mégahertz (un million de hertz), la valeur obtenue est donc exprimée en partie par million (ppm). Néanmoins cela suppose que l'on ait une substance de référence pour chaque noyau.
Pendant les années suivant l'apparition de la RMN en onde continue, il n'était pas réellement envisageable de faire autre chose que de la RMN du proton, grâce à sa sensibilité et son abondance naturelle. Dans la mesure où très peu de signaux dépassaient 10 ppm (voir ci-dessous), on a utilisé l'échelle τ (Tau) pendant 15-20 ans avec la relation : τ = 10 - δ. La référence pour le proton était, et est toujours, le tétramétylsilane (SiMe4) en interne (donc mélangé avec la substance).
À l'heure actuelle, on utilise exclusivement l'échelle δ.
Références
La référence choisie dépend de l'isotope étudié.
En RMN en solution
Par exemple pour le 1H, 13C et 29Si, le tétraméthylsilane (ou TMS) est choisi en général. Les références (relativement) inertes chimiquement peuvent être utilisées de façon interne, c'est-à-dire mélangées dans le solvant. C'est le cas du TMS qui est inerte, et facile à éliminer puisqu'il bout à 26,6 °C, à raison d'environ 0,03 %. Sinon, la référence est dite externe. Dans ce cas, on effectue un premier spectre avec le composé à étudier puis un second en insérant un capillaire rempli de la « référence externe ». Le second spectre permet de calibrer correctement le premier spectre. C'est notamment le cas pour la RMN du phosphore 31 dont la référence est l'acide phosphorique (H3PO4 à 85 %).
On peut également utiliser une référence secondaire, c'est-à-dire un composé dont le déplacement chimique est connu. Le plus souvent, il s'agit des protons résiduels du solvant deutéré pour le proton ou du signal du solvant s'il contient le noyau étudié. Cependant, cette méthode est moins fiable car le déplacement chimique des références secondaires internes varie avec la concentration et surtout le pH de la solution.
Le tableau suivant présente les références utilisées pour d'autres noyaux[5].
| Noyau | 1H | 13C | 31P | 19F | 29Si | 6Li & 7Li | 15N | 11B |
| Référence | TMS | TMS | H3PO4 85 % (ext) | CFCl3 | TMS | LiCl/H2O 1M (ext) | MeNO2 | BF3•Et2O (ext) |
En RMN du solide
| Noyau | 1H | 19F | 23Na | 27Al | 67Zn | 71Ga | 115In | 207Pb |
| Référence en solutions aqueuses (1 M) | TMS | CFCl3 | NaNO3 | Al(NO3)3 | Zn(NO3)2 | Ga(NO3)3 | In(NO3)3 | Pb(CH3)4 |
Les interactions dipolaires magnétiques

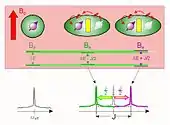
Les interactions dipolaires magnétiques entre deux spins couplés donnent lieu à une variation d'énergie de leurs états quantiques respectifs, et par conséquent modifient leurs résonances.
Cette interaction peut exister soit directement à travers l'espace (couplage dipolaire direct), soit par l'intermédiaire des électrons situés dans les orbitales moléculaires de liaison (appelé couplage dipolaire indirect, scalaire ou encore couplage J)[6].
Les couplages dipolaires directs
Le couplage dipolaire direct dépend de la distance entre deux noyaux et en interaction et de leurs rapports gyromagnétiques, et .





L'intensité du couplage (souvent appelée constante dipolaire) peut être définie (en Hz) par :

où est la perméabilité magnétique du vide.

Par exemple, deux protons séparés de 2Å présentent une constante dipolaire de -59,311 kHz[7].
Le déplacement des niveaux d'énergie produit un éclatement dipolaire, c'est-à-dire l'apparition d'un doublet à la place d'une résonance unique pour chacun des spins. La valeur de l'éclatement est donnée par :

où est l'orientation du vecteur internucléaire , c'est-à-dire reliant les deux noyaux, par rapport au champ appliqué .


vaut 3/2 si les deux noyaux sont identiques, par exemple deux protons (couplage homo-nucléaire). Il vaut 1 dans le cas de deux noyaux différents (couplage hétéro-nucléaire).

Si les réorientations moléculaires sont très rapides comme dans les liquides, l'éclatement disparait, car la moyenne s'annule.

Dans les solides, la mesure de l'interaction dipolaire est un moyen puissant d'obtenir les distances interatomiques[8].
Les couplages dipolaires indirects ou scalaires
Les couplages dipolaires directs (à travers l'espace) sont annulés dans les liquides, mais lorsqu'une liaison chimique existe les spins sont tout de même couplés via les électrons[9]. Ce couplage indirect qui résiste aux réorientations moléculaires est donc souvent appelé couplage scalaire (c'est-à-dire indépendant de l'orientation). Il se nomme aussi couplage « J » en spectroscopie RMN en référence à la constante qui en définit l'intensité (n indique le nombre de liaisons séparant les deux spins i et j).

Lorsque la différence de déplacement chimique en hertz (Δν) est supérieur à 5-10 fois la constante de couplage J (en valeur absolue), le couplage est dit du premier ordre. En dessous de cette limite, le couplage est dit du second ordre et cela peut compliquer notablement l'interprétation.
Le couplage dipolaire scalaire est un excellent indicateur de la présence de liaison chimique et une aide précieuse dans la détermination des structures de molécules organiques.
L'interaction quadripolaire
L'interaction quadripolaire[10] ne concerne que les isotopes atomiques de spin égal ou supérieur à 1. Ceux-ci représentent environ les 3/4 des isotopes observables en RMN, parmi lesquels des atomes très importants en chimie comme l'oxygène (17O), l'azote (14N) et le chlore (35Cl, 37Cl).
La principale caractéristique qui les distingue des noyaux de spin 1/2 comme l'hydrogène (1H) ou le carbone (13C) est la présence d'un moment quadripolaire électrique[11] en plus du moment magnétique nucléaire. Ce moment est dû à une distribution anisotrope des charges dans le noyau. Ce moment quadripolaire électrique est susceptible d'interagir avec tout gradient de champ électrique non nul au niveau du noyau. Cette interaction quadripolaire est susceptible de modifier très fortement l'énergie des états quantiques et donc influence la fréquence de résonance et la relaxation.
Les effets sur la fréquence de résonance disparaissent dans les liquides à cause des réorientations rapides des molécules. L'interaction quadripolaire devient nulle également si le noyau dans un solide est localisé dans un environnement très symétrique (symétrie sphérique du gradient de champ électrique).
L'anisotropie des interactions

Dans la majeure partie des cas, les électrons n'ont pas une distribution sphérique autour du noyau. Cette distribution dépend beaucoup de la géométrie de la molécule, il en résulte que les interactions internes sont en général anisotropes.
Dans les solutions liquides, ce n'est pas important, car on n'observe qu'une valeur moyenne à cause des rapides réorientations moléculaires (mouvement brownien). Dans les solides par contre, la position des raies de résonance est alors fortement modifiée suivant l'orientation de la molécule, du cristal ou plus généralement de la cristallite.
Dans le cas du déplacement chimique par exemple, la constante d'écran ne sera plus représentée par un simple scalaire , mais par un tenseur du second ordre, qui définit les composantes de la constante d'écran selon les différentes directions de l'espace. Il existe toujours trois directions particulières orthogonales, c'est-à-dire un système d'axes principaux, qui peuvent être choisies comme référentiel pour décrire l'interaction de telle sorte que le tenseur soit diagonal dans ce système d'axes. Ainsi :


Les trois scalaires , et , correspondent à la valeur de la constante d'écran selon les trois directions principales. Ce tenseur peut se représenter sous forme d'un ellipsoïde dont la forme dépend de la symétrie de l'interaction. Notamment, si l'interaction est de symétrie axiale (), l'ellipsoïde est un ellipsoïde de révolution. Si les trois valeurs sont égales, le système est isotrope et peut alors être décrit par une seule constante d'écran .





La fréquence de résonance de chaque noyau dépendra directement du tenseur d'écran et pourra s'écrire :

où les angles et sont les coordonnées angulaires sphériques donnant l'orientation de l'axe principal par rapport au champ magnétique , et est la fréquence de Larmor,



Les échantillons solides sont souvent analysés sous forme de poudre, c'est-à-dire qu'ils contiennent un ensemble de cristallite dont les orientations sont aléatoires. Le spectre de résonance observé correspondra alors à la superposition des résonances de toutes les cristallites présentes. Un tel spectre est appelé spectre de poudre et présente des formes caractéristiques de la symétrie du tenseur d'écran.
L'anisotropie de déplacement chimique est en général désignée par l'acronyme anglais CSA (pour chemical shift anisotropy).
L'interaction dipolaire est aussi anisotrope dans les solides, mais a la particularité d'être de symétrie axiale.
Autres interactions possibles dans les substances paramagnétiques
Bien que la RMN soit généralement appliquée sur des substances diamagnétiques, il n’est pas impossible de l’utiliser pour des substances paramagnétiques. Dans ce cas, il y a des effets particuliers dont il faut tenir compte car ils peuvent déplacer les résonances ou les élargir si fortement qu’il devient souvent difficile de les détecter.
Déplacement de Knight
Le déplacement de Knight K caractérise la fréquence RMN de noyaux atomiques dans un métal (par exemple le sodium) comparée à celle des mêmes noyaux dans un environnement non-métallique (par exemple, le sodium dans NaCl). Le déplacement observé reflète les champs magnétiques locaux produits sur les noyaux par l’aimantation des électrons de conduction. Ce déplacement généralement anisotropique peut être de l’ordre de milliers de ppm. Les déplacements chimiques des substances non-métalliques sont négligeables par comparaison.
Déplacement de contact et pseudocontact
Le spin des électrons non-appariés influence les résonances RMN de deux façons, par déplacement de contact (contact shift) ou de pseudo contact (pseudo-contact shifts). Les deux effets sont simultanés mais l’un d’eux peut être parfois prédominant. Le déplacement de contact résulte de la polarisation des spins électroniques transmise au travers des orbitales moléculaires. Le déplacement de pseudocontact résulte des champs magnétiques locaux produits par les centres paramagnétiques (c’est un couplage dipolaire électrons-noyaux qui varient donc en 1/r3).
Comme exemple, la résonance des protons dans le nickelocène est à environ -255 ppm, alors que dans le composé analogue diamagnétique, le ferrocène, elle se situe vers 5 ppm[12].
Réglages du spectromètre
Même si le présent article porte principalement sur la théorie de la RMN, il est important de comprendre comment la théorie est mise en pratique au niveau du spectromètre.
Pour chaque échantillon, il faut :
- stabiliser le champ magnétique (le lock) ;
- régler l'homogénéité du champ magnétique (le shim).
Le lock
Le premier cas est appelé le lock en anglais (« verrouillage » en français mais ce terme est très rarement utilisé). Il s'agit de trouver une fréquence par rapport à laquelle on peut déterminer que le champ magnétique varie. Ce dernier varie inévitablement parce que l'aimant supraconducteur se décharge mais cela est un processus très lent. En revanche, n'importe quel objet ou personne en mouvement dans le champ magnétique va créer des variations brutales qui doivent être compensées rapidement.
En cas de variation, la fréquence d'excitation des différents noyaux est ajustée. Le moyen le plus simple de trouver une fréquence de référence est d'utiliser le signal du deutérium du solvant ; on utilise un solvant deutéré pour dissoudre les composés et la présence de deutérium en grande quantité est une aubaine. Le spectromètre effectue un balayage de fréquences pour observer le déplacement du signal du deutérium et répercute cette différence vers tous les noyaux.
Le shim
En anglais, le terme « shim » désigne une sorte de cale. À l'origine de la RMN, il fallait en effet placer des cales à certains endroits pour homogénéiser le champ magnétique. Aujourd'hui, on a ajouté des électro-aimants capables de générer des champs magnétiques de différentes formes pour compenser les inhomogénéités de l'aimant supraconducteur, mais le terme est resté.
Le verbe anglicisé « shimmer » est donc devenu courant en RMN et, par extension, dans les domaines utilisant la RMN. On peut donc dire d'un spectre qu'il est bien/mal « shimmé ». Lorsque le spectre est "mal shimmé", les signaux observés ne sont pas symétriques. Des trainées à droite ou à gauche du signal sont alors observables.
Le réglage du shim peut se faire :
- manuellement : puisque le lock utilise le signal du deutérium (en dispersion), on peut également utiliser ce signal pour shimmer l'échantillon. Le signal en absorption est facilement détecté et ce signal est de plus en plus fin, et donc haut (puisque la surface est constante), lorsque l'homogénéité du champ s'améliore. Le réglage itératif du champ en z, z2 et z3 avec rotation du tube permet de faire les derniers réglages du shim ;
- automatiquement :
- sur les anciens spectromètres, le shim était effectué comme un shim manuel,
- sur les nouveaux spectromètres Bruker, et avec la sonde adéquate, le shim automatique (seulement des bobines en Z) se fait par un programme appelé topshim qui utilise les gradients de champ pour déterminer les valeurs optimales des différents réglages. L'efficacité est d'autant meilleure que le nombre d'atomes de deutérium est élevé dans le solvant et que le champ est fort. Ceci peut poser problème avec du chloroforme deutéré sur un spectromètre de 300-400 MHz.
Séquences d'impulsion

La technique de base pour obtenir un spectre RMN consiste en l'application d'une impulsion de champ radiofréquence selon l'axe x ou y. Cette impulsion a une durée (déterminée expérimentalement) permettant d'obtenir le signal maximum dans la bobine de détection, c'est-à-dire en faisant basculer le vecteur d'aimantation dans le plan xy perpendiculairement à l'axe z (qui correspond à la direction du champ magnétique B0). Pour cette raison, on appelle ce genre d'impulsion une impulsion à/de 90°. À la suite de cette impulsion, l'aimantation transverse dans le plan xy est mesurée et convertie en données numériques grâce à un convertisseur analogique-numérique tandis qu'elle diminue du fait de la relaxation. Trois types de relaxation peuvent être différenciées :
- T1, appelé le temps de relaxation spin-réseau. C'est le temps nécessaire pour le retour à l'équilibre thermodynamique des spins qui ont été excités par l'impulsion radio-fréquence. Par définition, comme la relaxation spontanée est négligeable en RMN, cette relaxation implique un échange d'énergie avec l'environnement des noyaux (le « réseau »), d'où son nom de relaxation spin-réseau. La valeur de T1 est de l'ordre de la seconde et correspond au temps nécessaire pour que 63 % des spins soient revenus à leur état fondamental. Elle dépend de la molécule et du noyau considéré.
- T2, appelé le temps de relaxation spin-spin. C'est un phénomène assez complexe, de nature entropique, qui correspond à la perte de cohérence des spins responsables de l'aimantation transverse, c'est-à-dire dans le plan xy. Les fluctuations des champs magnétiques locaux dans l'échantillon induisent des modifications aléatoires des vitesses de précession des différents spins. Le résultat est que la cohérence de phase des spins initialement produite par l'impulsion est progressivement perdue, jusqu'à ce qu'il n'y ait plus aucune cohérence et que l'aimantation dans le plan xy devienne nulle. Ceci se traduit par un élargissement des pics d'autant plus important que ces déphasages sont rapides.
- T2* : une raison pour laquelle les spins peuvent se déphaser est l'inhomogénéité du champ magnétique B0. Lorsque le champ n'est pas assez homogène, les spins, qui sont en mouvement permanent dans l'échantillon, vont alterner entre des zones de champ plus fort ou plus faible. Ceci provoque, comme le T2, un déphasage des spins qui crée au mieux un élargissement des pics mais, éventuellement, une annulation du signal. T2* est en fait la somme du T2 réel et du Tinhom, dû à l'inhomogénéité du champ.
Le signal mesuré (une acquisition, un scan en anglais) est normalement appelé le signal de précession libre, mais plus souvent appelé la FID (d'après l'anglais Free Induction Decay) et se présente comme une somme de sinusoïdes apparemment indéchiffrable. En fait, il ne s'agit pas d'une seule mesure mais d'une moyenne d'acquisitions effectuées l'une après l'autre. Ceci permet d'augmenter l'intensité des vrais signaux par rapport au bruit de fond (aléatoire) qui se moyenne autour de zéro, c'est-à-dire que l'on augmente le rapport signal/bruit ; pour n acquisitions, on augmente le rapport signal/bruit de la racine carré de n. La conséquence pratique est que pour augmenter le rapport signal/bruit d'un facteur 4, il va en fait falloir multiplier le nombre d'acquisitions (et donc le temps) par 16, soit 42. La décroissance apparente de la FID est due à la relaxation T1 et T2*, mais aussi à des effets expérimentaux liés par exemple à l'effet de molécules paramagnétiques dans l'échantillon, comme l'oxygène gazeux.
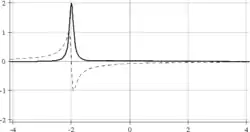
Afin de connaître le signe des fréquences de résonance, il est nécessaire de faire une détection synchrone en quadrature, c'est-à-dire la mesure de deux signaux décalés temporellement d'un quart de période de Larmor (traits plein et pointillé sur la figure ci-dessous). Chaque point de la FID est donc en fait un nombre complexe.

Après un éventuel traitement mathématique du signal, une transformée de Fourier est appliquée pour extraire la fréquence de chaque sinusoïde de la FID et produire un spectre RMN. Le signal en ligne continue est appelé le signal en absorption alors que celui en pointillé est appelé le signal en dispersion (il y a une différence de phase de 90° entre les deux).
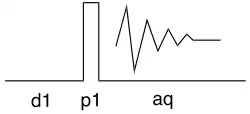
La séquence d'impulsion la plus simple est représentée ci-dessous : après un délai d1, le spectromètre génère une impulsion forte à 90° (p1, p pour pulse en anglais) puis effectue l'acquisition du signal (aq). La raison pour laquelle on ajoute le délai d1 est que cette séquence est répétée un certain nombre de fois et qu'il nécessaire, entre deux impulsion-acquisition, que le système se remette à l'équilibre.
Si la séquence d'impulsion la plus simple se résume à une seule impulsion suivie de la mesure de la FID résultante, le développement d'impulsions plus sophistiquées est un des enjeux méthodologique de la spectroscopie RMN moderne. Des enchainements d'impulsions sur plusieurs noyaux permettent une manipulation à volonté des spins nucléaires; une extension importante de ces séquences d'impulsion est la spectroscopie 2D (à deux dimensions) et même nD (multidimensionnelle) qui permettent en particulier d'établir des corrélations entre différentes résonances.
Cycles de phase
Afin de supprimer les défauts dus à l'électronique, à des radiofréquences parasites ou pour supprimer certains artéfacts, on alterne les impulsions et la détection selon les axes x, -x, y et -y. Ceci s'appelle le cycle de phase. L'addition ou la soustraction des différentes FID selon leur phase (positive ou négative) permet de supprimer les artéfacts qui, eux, conservent le même signe.
Pour un simple spectre du proton, on utilise en général un cycle de phase en huit étapes :
- impulsion : x, x, -x, -x, y, y, -y, -y ;
- détection : x, x, -x, -x, y, y, -y, -y
donc le premier scan s'effectue avec une impulsion selon l'axe x et la détection selon x. Le deuxième scan est identique. Le troisième s'effectue avec une impulsion en -x et une détection en -x également, etc.
La principale conséquence de ce cycle de phase est que le nombre d'acquisitions effectuées devrait toujours être un multiple du nombre d'étapes dans le cycle de phase.
Temps d'acquisition

Le temps d'acquisition (AQ) ne peut pas être modifié directement. Il dépend de deux paramètres importants :
- le nombre de points utilisés pour numériser la FID ;
- la gamme spectrale (exprimée en Hz) à numériser.
- secondes.

Un temps d'acquisition trop long ne représente pas de problème, mis à part une perte de temps. En revanche, un temps d'acquisition trop court va tronquer la FID avant que celle-ci ne soit revenue à zéro. Ceci provoque une perte du signal, l'apparition d'artéfacts après la transformée de Fourier et, éventuellement, la perte des petites constantes de couplage (qui mettent plus de temps à se développer). Voir le schéma ci-contre.
Délai de relaxation
Le délai de relaxation, à ne pas confondre avec le temps de relaxation, correspond à la période de temps nécessaire pour que tous les spins soient revenus à leur état fondamental. Il correspond à 5 * T1, où T1 est le temps de relaxation spin-réseau le plus long de la molécule.
Traitement du signal
Si la transformée de Fourier, déjà citée ci-dessus, est la base de traitement du signal en RMN, il y a beaucoup d'autres traitements mathématiques importants. Ces traitements sont décrits ci-dessous dans l'ordre habituellement utilisé.
Les nombres correspondant à un nombre de points sont généralement exprimés en « kilo » dans le sens informatique du terme, c'est-à-dire que 1 k = 1 024 valeurs car il s'agit de la puissance de 2 la plus proche de 1 000 (210).
Taille de la FID et résolution
Comme indiqué ci-dessus dans Temps d'acquisition, il faut indiquer la gamme spectrale (en Hz) à numériser ainsi que le nombre de points.
Selon le spectromètre, le nombre de points peut en fait correspondre au nombre de valeurs réelles et imaginaires, ce qui signifie que le nombre de valeurs complexes est en fait la moitié du nombre de points : une FID de 64 k va donc être un fichier de 32 k de valeurs réelles et 32 k de valeurs imaginaires. Au total, cela représente 32 k valeurs complexes.
La résolution sera donc la gamme spectrale divisée par le nombre de points réels : par exemple, 20 ppm en proton à 400 MHz représentent 8 000 Hz (20×10−6×400×106). Si l'on choisit un nombre de points de 64 k, cela représente une résolution de 0,24 Hz/point (20×400/2). Cela est très correct en proton.
Le zero-filling
Afin d'améliorer la résolution, on applique souvent un zero-filling (le terme anglais), c'est-à-dire que l'on rajoute à la FID un certain nombre de points d'une valeur nulle ; en général, on double le nombre total de points. On utilise ensuite une multiplication exponentielle (voir Convolutions / fenêtrage ci-dessous) pour être sûr de ne pas avoir de troncature du signal. Ceci permet d'améliorer la résolution du signal en la divisant par deux. Cependant, il ne faut pas espérer révéler des couplages qui n'ont pas été détectés pendant l'acquisition.
Ligne de base de la FID
La FID est une somme de sinusoïdes qui devraient être centrées sur zéro selon l'axe y. Un décalage vertical est possible et il peut être compensé par le logiciel de traitement RMN.
Apodisation
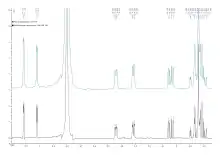
Un certain nombre de fonctions mathématiques appliquées à la FID (apodisation ou fenêtrage) vont avoir des effets très importants sur le spectre obtenu. Parmi tant d'autres :
- EM : Exponential Multiplication en anglais. On multiplie la FID par une exponentielle inverse ce qui va annuler le signal en fin d'acquisition. Les pics les plus importants sont donc conservés alors que les signaux tardifs (le bruit de fond) est atténué, ce qui est particulièrement conseillé en cas de zero-filling. Cela permet d'augmenter notablement le rapport signal/bruit mais au détriment de la résolution car les pics sont élargis. Le seul paramètre est nommé LB (Line Broadening). Une valeur de 0,15 Hz est généralement employée en proton ou 1/100e de la résonance du proton en 13C ou en 31P (soit 4 Hz pour un spectromètre à 400 MHz). C'est une des rares fonctions qui n'altère pas la possibilité d'intégrer les signaux ;
- amélioration de la résolution : certaines fonctions permettent, selon la valeur de leurs paramètres, d'augmenter la résolution du spectre au détriment du rapport signal/bruit. On peut citer la multiplication gaussienne (GM), le sinus (SIN) ou le sinus carré (QSIN). Voir ci-dessous.
Transformée de Fourier
La vraie transformée de Fourier n'est pas applicable en RMN car elle requiert une intégration de -∞ à +∞. On utilise donc ce que l'on appelle une transformée de Fourier discrète et, plus encore, une transformée de Fourier rapide qui est un algorithme très rapide, notamment si le nombre de valeurs est une puissance de 2. Ceci explique l'utilisation du « kilo » dans le sens informatique du terme : une puissance de 2 ().

Phasage

Dans un spectre RMN simple, un signal est dit « phasé » lorsque son profil est positif et symétrique à gauche et à droite. Notamment, le signal doit s'élargir de la même façon à gauche et à droite pour se fondre avec la ligne de base.
Plusieurs phénomènes font que les signaux détectés sont déphasés par rapport au cas idéal :
- tous en même temps autour de l'axe δ (l'ordre zéro)
et
- les uns par rapport aux autres en fonction du déplacement chimique (l'ordre un).
Cette étape est assurée par le logiciel de traitement RMN de façon interactive avec l'utilisateur.
Étalonnage
L'étalonnage s'effectue :
- en attribuant une valeur connue à la référence interne ou à une référence secondaire interne comme les signaux du solvant deutéré (ou des restes de solvant protoné) ;
- en menant une autre expérience avec une référence externe. Le décalage entre le signal mesuré et la référence est alors reporté dans le premier spectre.
Si cela n'est pas possible, un étalonnage par défaut est réalisé par le logiciel d'acquisition.
Détection des pics (peak-picking)
Une fois la phase correctement ajustée pour tous les pics, le logiciel de traitement RMN peut détecter les pics (positifs ou négatifs) par leur extremum. On utilise souvent le terme anglais peak picking pour cette étape.
Correction de la ligne de base
Dans le cas idéal, toutes les expériences de RMN devraient aboutir à une ligne de base plane et centrée sur l'axe δ mais l'on n'est presque jamais dans ce cas. Les différentes méthodes de correction sont :
- le décalage (offset en anglais) : on centre la moyenne du bruit de fond à 0 sur l'axe y ;
- l'ajustement polynomial : on ajuste les valeurs selon un polynôme d'ordre variable ;
- le spline cubique : l'utilisateur indique les points devant être ajustés à la valeur zéro sur l'axe y. Une interpolation (bi)cubique est faite entre ces points et permet de corriger la ligne de base.
Intégration des signaux
L'intégration est une simple addition des intensités des signaux entre deux valeurs de δ. Pour qu'elle soit bien faite, une intégration doit être effectuée entre -5 et +5 fois la largeur à mi-hauteur du pic considéré. Le résultat doit être une courbe qui commence et se termine par une ligne horizontale.
La RMN en chimie organique

Dans la mesure où la chimie organique est la chimie du carbone ET de l'hydrogène, la spectroscopie RMN est un des outils d'analyse les plus utilisés en chimie organique.
Les noyaux les plus souvent étudiés sont l'1H, le 13C, le 31P, le 15N et le 19F de spin nucléaire égal à 1/2. Sont étudiés également l'17O de spin 5/2 et le 14N de spin 1.
L'échantillon à analyser est généralement mis en solution dans un solvant, souvent du chloroforme deutéré, (CDCl3) contenant éventuellement un petit pourcentage de tétraméthylsilane. Ce solvant est largement enrichi en deutérium (2D, un isotope de l'hydrogène) afin qu'il soit invisible en RMN du proton de sorte que l'on n'observe en RMN que la petite fraction (env 0,2 %) de CHCl3 restante.
La réalisation d'un spectre RMN du 1H est généralement rapide (quelques minutes d'acquisition). Grâce à l'analyse des déplacements chimiques de chaque résonance et de leur structure fine (multiplet) due aux couplages scalaires, il est possible de déterminer la structure de beaucoup de molécules organiques. Cette détermination est facilitée en utilisant aussi en parallèle la spectroscopie RMN du 13C. L'acquisition d'un spectre du 13C est cependant plus longue compte tenu de l'abondance naturelle très faible de cet isotope du carbone (~ 1,1 %).
Les techniques de spectroscopie RMN multidimensionnelle sont utilisées pour faciliter l'interprétation de spectres complexes.
L'interprétation des spectres RMN simples
Les spectres RMN sont d'une complexité qui dépend de la molécule et du noyau étudié. Dans la majorité des cas en chimie organique, on va se baser sur la RMN du proton et la RMN du carbone 13. Si les spectres sont relativement simples, on peut attribuer les signaux aux différents noyaux en utilisant :
- Le déplacement chimique (δ) : des tables[13] donnent des gammes de déplacements chimiques pour les différents noyaux selon les groupes fonctionnels environnant.
- Les constantes de couplages.
- La multiplicité des pics : sauf pour le 13C (en général), la multiplicité du signal en 1H, 19F, 31P donne des informations importantes sur le nombre de spins voisins.
- L'intégration des pics : sous réserve que l'expérience permette l'intégration des pics, cette intégration est proportionnelle au nombre de spins.
Si la structure n'a pas pu être complètement résolue, d'autres expériences simples peuvent être menées :
- Le DEPT en 13C permet de différencier les groupes C, CH, CH2 et CH3 très rapidement (le DEPT est environ quatre fois plus rapide que l'acquisition d'un spectre 13C).
Voir :
Spectroscopie RMN de corrélation
La spectroscopie de corrélation[14] est un type de spectroscopie 2D (bidimensionnelle) ou parfois nD (multidimensionnelle). Le tableau suivant donne quelques exemples de ce type de spectroscopie. La spectroscopie homonucléaire concerne un seul type d'isotopes et la spectroscopie hétéronucléaire concernent deux isotopes différents.
| Nombre de dimension | Spectroscopie | Acronyme | Nom complet en anglais |
|---|---|---|---|
| RMN 2D | Homonucléaire | COSY | Correlation spectroscopy |
| NOESY | Nuclear Overhauser effect spectroscopy | ||
| TOCSY | Total correlation spectroscopy | ||
| Hétéronucléaire | HMQC | Heteronuclear multiquantum coherence | |
| HSQC (en) | Heteronuclear Single Quantum Coherence | ||
| HMBC | Heteronuclear Multi-Bond Connectivity | ||
| HOESY | Heteronuclear Overhauser Effect Spectroscopy | ||
| RMN 3D (en) | Hétéronucléaire | HNCA (en) | Amide proton to Nitrogen to C α-carbon correlation |
| HNCOCA (en) |
Les spectres à multiples dimensions donnent plus informations que les spectres classiques à une dimension et sont particulièrement utiles pour déterminer la structure d'une molécule, particulièrement celles qui sont trop complexes à étudier à l'aide la spectroscopie 1D.[réf. souhaitée]
La RMN en biologie structurale
À côté de la radiocristallographie, la RMN est devenue une méthode d'étude des macromolécules biologiques en solution. Elle ne nécessite pas l'obtention de monocristaux et permet d'étudier des protéines, des acides nucléiques à des concentrations millimolaires. Les techniques de RMN multidimensionnelles conduisent à corréler les fréquences de plusieurs spins et de résoudre les ambiguïtés liées aux superpositions spectrales. Des protéines de masse moléculaire de 10 à 30 kDa peuvent être analysées ainsi que des oligonucléotides de plusieurs dizaines de paires de bases.
RMN homonucléaire sans marquage isotopique
Historiquement, les protéines ont été étudiées par la RMN du proton (isotope 1H) présent en abondance.
Une première étape consiste à attribuer les résonances, c'est-à-dire à établir une corrélation entre les signaux du spectre et les atomes d'hydrogène de la molécule. Deux expériences clefs sont utilisées, l'expérience de corrélation par les couplages scalaires (HOHAHA ou TOCSY) et l'expérience de corrélation à travers l'espace par effet Overhauser (NOESY). Cette attribution est dite séquentielle, car elle opère par positionnement relatif d'un noyau par rapport à ses voisins en utilisant l'information de la séquence peptidique. Ce processus d'attribution (semblable à un puzzle) devient de plus en plus complexe à mesure que la taille de la protéine augmente; de plus, l'effet Overhauser se transmet à travers l'espace et ne permet pas de distinguer des noyaux proches dans la séquence peptidique et ceux proches dans l'espace. Des erreurs sont donc possibles qui ne se détectent qu'à la fin du processus, lorsque certaines pièces du puzzle restent sur le tapis.
Une fois les spectres attribués, les informations sont alors utilisées de manière quantitative : les couplages scalaires renseignent sur les angles dièdres et les effets Overhauser sur des distances interatomiques (jusqu'à 4-6 angströms). Ces informations sont introduites dans des programmes de modélisation moléculaire, pour rechercher une ou plusieurs conformations de la molécule compatibles avec les données. La stratégie est comparable à celle du géomètre qui mesure des distances et des angles entre bâtiments et recalcule un plan de ville. Sauf que la portée des distances mesurées en RMN est faible par rapport aux objets évalués; l'accumulation des erreurs expérimentales et/ou le faible nombre de données conduisent à des structures localement mal définies.
Deux difficultés limitent la taille des macromolécules accessibles à la technique : la complexité des spectres et la largeur individuelle de chaque signal. Si la taille de la protéine double, le nombre de résonances du spectre va doubler sans que la dispersion (c'est-à-dire la largeur spectrale) n'augmente. Une solution consiste à recourir à un spectromètre RMN à plus haut champ et donc beaucoup plus coûteux. Si la taille double, la masse moléculaire augmente et la protéine tourne plus lentement sur elle-même par mouvement diffusif. Cela conduit à des signaux plus larges, car la relaxation transversale devient plus performante. L'augmentation du champ magnétique est sans effet et il faut trouver une autre parade au problème (réduction de la viscosité, réduction du nombre de protons voisins…).
RMN hétéronucléaire avec marquage isotopique
Afin de résoudre les superpositions spectrales dans les grosses molécules, il s'avérait nécessaire de passer de la RMN 2D à la RMN 3D. Des essais peu fructueux ont été faits au début des années 1990 pour combiner les séquences HOHAHA et NOESY dans une expérience tridimensionnelle. Si on dispose d'une protéine entièrement enrichie en 15N et en 13C, on peut concevoir des expériences de corrélation entre spins, uniquement fondées sur des couplages scalaires et permettant de relier tout le squelette peptidique ainsi que les chaînes latérales. Les isotopes naturellement abondants sont respectivement le 14N (noyau quadripolaire) et le 12C (noyau invisible en RMN), mais la plupart des protéines étant obtenues par surexpression bactérienne, il est possible de faire des cultures sur des milieux enrichis isotopiquement.
Cette nouvelle stratégie fait appel à une série d'expériences « 3D triple résonance » : 3D car un spectre tridimensionnel est obtenu, triple résonance car les fréquences de trois noyaux différents sont détectées. Pour des raisons de sensibilité, toutes ces séquences partent du 1H (noyau au rapport gyromagnétique élevé) et se terminent par la détection du même noyau. Dans l'expérience HNCO, on établit donc une corrélation entre le proton amide (HN), son azote (N) et le carbonyle (CO) de l'acide aminé précédent. Les couplages scalaires utilisés pour les transferts de cohérence sont des couplages 1J (à une liaison) et donc relativement importants (1JNH = 90 Hz et 1JNCO = 15 Hz). Cela assure donc une grande efficacité de transfert même dans le cas de protéines de masse élevées (largeur de raie). À noter que cette expérience HNCO permet de relier par couplage scalaire des acides aminés, ce que la stratégie homonucléaire ne permettait pas (absence de couplage scalaire 3J à travers la liaison peptidique).
La RMN des solides
La rotation à l'angle magique
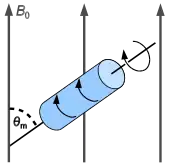
Dans les liquides, les mouvements browniens entrainent des réorientations très rapides de molécules, de sorte que seule la moyenne des interactions prise sur l'ensemble des orientations est mesurée. Dans les solides, ce n'est pas le cas puisque les mouvements des molécules sont souvent beaucoup plus lents que la mesure du signal RMN. Les spectres de poudres sont parfois très larges si le milieu est très anisotrope ; les spectres se superposent et la séparation d'espèces chimiques différentes en fonction de leur déplacement chimique par exemple n'est donc pas aussi facile que dans les liquides.
Dans les années 1960, Andrew[4] a montré cependant qu'il était possible d'obtenir des spectres correspondant à la moyenne isotrope des interactions en faisant tourner l'échantillon autour d'un axe incliné de arccos(1/√3) ≈ 54° 44' par rapport au champ magnétique. Cet angle est appelé, l’angle magique, et la technique de rotation à l'angle magique est en général désignée par son acronyme anglais : MAS (pour magic angle spinning)
Une condition essentielle pour que cette technique MAS soit réellement efficace est que la vitesse de rotation soit au moins de l'ordre de grandeur de l'amplitude de l'anisotropie. Ainsi pour moyenner efficacement l'interaction dipolaire entre un atome de 13C et un proton, la vitesse ou plutôt la fréquence de rotation doit être de l'ordre de 30 kHz (c'est-à-dire 30 000 tours par seconde). Ce sont des vitesses très importantes qui ne peuvent être obtenues qu'en plaçant l'échantillon dans de petits conteneurs cylindriques (le rotor) placés en rotation dans une turbine à coussins d'air. Les vitesses actuellement atteintes avec ces systèmes avoisinent les 90 kHz.
Notes et références
Notes
- Pour des raisons historiques, l’axe des fréquences en RMN est orienté généralement de droite à gauche, c’est-à-dire à l’opposé du « blindage ».
Références
- (en) E.L. Hahn, « Spin Echoes », Physical Review, vol. 80, no 4, , p. 580 (DOI 10.1103/PhysRev.80.580).
- (en) W. C. Dickinson, « Dependence of the 19F Nuclear Resonance Position on Chemical Compound », Physical Review, vol. 77, no 5, , p. 736 (DOI 10.1103/PhysRev.77.736.2).
- (en) W.G. Proctor, F.C. Yu, « The Dependence of a Nuclear Magnetic Resonance Frequency upon Chemical Compound », Physical Review, vol. 77, no 5, , p. 717 (DOI 10.1103/PhysRev.77.717).
- (en) E. R. Andrew, A. Bradbury et R. G. Eades, « Removal of Dipolar Broadening of Nuclear Magnetic Resonance Spectra of Solids by Specimen Rotation », Nature, vol. 183, no 4678, , p. 1802-1803 (DOI 10.1038/1831802a0).
- Bruker Biospin Almanac, 2007, p. 23.
- Abragam 1994.
- Levitt 2008.
- T. Gullion et J. Schaefer, « Rotational-echo double-resonance NMR », Journal of Magnetic Resonance, vol. 81, no 1, , p. 196-200 (DOI 10.1016/0022-2364(89)90280-1).
- Norman F. Ramsey, « Electron Coupled Interactions between Nuclear Spins in Molecules », Phys. Rev., vol. 91, no 2, , p. 303 (DOI 10.1103/PhysRev.91.303).
- (en) A. Jerschow, « From nuclear structure to the quadrupolar NMR interaction and high-resolution spectroscopy », Progress in Nuclear Magnetic Resonance Spectroscopy, vol. 46, no 1, , p. 63-78.
- (en) P. Pyykkö, « Year-2008 nuclear quadrupole moments », Molecular Physics, vol. 106, no 16, , p. 1965-1974.
- Henrike Heise, Frank H Köhler et Xiulan Xie, « Solid-State NMR Spectroscopy of Paramagnetic Metallocenes », Journal of Magnetic Resonance, vol. 150, no 2, , p. 198–206 (ISSN 1090-7807, DOI 10.1006/jmre.2001.2343, lire en ligne).
- Bruker, Almanac 2007, Bruker Biospin.
- (en-US) vickyh, « Spectrum Descriptions », sur www.protein-nmr.org.uk (consulté le ).
Bibliographie
Ouvrages de base recommandés
- (en) M. H. Levitt, Spin Dynamics : Basics of Nuclear Magnetic Resonance, Wiley, , 2e éd. (ISBN 978-0-470-51117-6)
- Daniel Canet, La RMN : concepts et méthodes, Paris, Interéditions, , 274 p. (ISBN 978-2-7296-0375-5)
- (en) James Keeler, Understanding NMR Spectroscopy, Chichester, Wiley, , 476 p., poche (ISBN 978-0-470-01787-6, LCCN 2005022612)
Généraux
- (en) A. Abragam, The Principles of Nuclear Magnetism, Oxford, Oxford University Press, , 599 p., poche (ISBN 978-0-19-852014-6, LCCN 61002889, lire en ligne), La « bible » (difficile)
- (en) C. P. Slichter, Principles of Magnetic Resonance (Springer Series in Solid-State Sciences), Berlin, Springer, , 3e éd., 658 p. (ISBN 978-3-540-50157-2, lire en ligne)
- (en) M. Mehring, Principles of High Resolution Nmr in Solids, Berlin, Springer-Verlag, , 2e éd., 344 p. (ISBN 978-0-387-11852-9, LCCN 82010827)
- (en) Richard R. Ernst, Principles of Nuclear Magnetic Resonance in One and Two Dimensions, Oxford, Oxford University Press, USA, , 610 p., poche (ISBN 978-0-19-855647-3, LCCN 85026014)
- (en) Eiichi Fukushima et Stephen B. W. Roeder, Experimental Pulse NMR : A Nuts and Bolts Approach, Reading, Westview Press, , 7e éd., poche (ISBN 978-0-201-62726-8, LCCN 81007901)
RMN en chimie organique
RMN en biochimie
- (en) John Cavanagh, Protein NMR Spectroscopy, Second Edition : Principles and Practice, Amsterdam, Academic Press, , 2e éd. (ISBN 978-0-12-164491-8, LCCN 2006021096)
- (en) Gordon S. Rule et T. Kevin Hitchens, Fundamentals of Protein NMR Spectroscopy, Dordrecht, Springer, , 1re éd., 532 p. (ISBN 978-1-4020-3499-2, LCCN 2008447860, lire en ligne)
RMN du solide
- (en) Melinda J. Duer, Introduction to Solid-State NMR Spectroscopy, Malden, Wiley-Blackwell, , 1re éd., 368 p., poche (ISBN 978-1-4051-0914-7, LCCN 2003063027)
- (en) Robin K. Harris, NMR Crystallography, Chichester, Wiley, , 520 p. (ISBN 978-0-470-69961-4, LCCN 2009031427, lire en ligne)
- (en) Klaus Schmidt-Rohr, Multidimensional Solid-State NMR and Polymers, Londres, Academic Press, , 478 p. (ISBN 978-0-12-626630-6, lire en ligne)
Voir aussi
Liens externes
- (en) Recommendations IUPAC 1997
- (en) NMR Wiki (Open NMR project), un wiki consacré à la RMN
- (en) NMR Predictor, un simulateur de spectres RMN (1H et 13C)
 Portail de la physique
Portail de la physique  Portail de la chimie
Portail de la chimie