Xanthogranulome juvénile
Le xanthogranulome juvénile (Juvenile xanthogranuloma ou JXG) est une forme de tumeur histiocytaire[1]. Elle est plus fréquemment présente sur la peau du bébé ou du jeune enfant sous forme de nodules composés de cellules histiocytaires, mais se développant parfois dans les yeux ou des organes internes (viscères)[2].
Cette lésion est généralement bénigne et évolue alors vers l'auto-guérison, mais peut poser problème quand elle se développe dans l'œil (risque de glaucome juvénile[3]) ou des viscères.
| Spécialité | Hématologie |
|---|
| CIM-10 |
D76.3 (ILDS D76.390) |
|---|---|
| DiseasesDB | 32580 |
| eMedicine | oph/588 |
| MeSH | D014972 |
![]() Mise en garde médicale
Mise en garde médicale
Il existe aussi un xanthogranulome de l'adulte[4] qui peut être bénin et guérir spontanément[5] ou très rarement présenter des formes nécrobiotiques de xanthogranulomes[6],[7],[8].
Classification
C'est l'une des formes d'histiocytoses non langerhansiennes (HCNL ou Non-X histiocytoses pour les anglophones) qui constitue elle-même la seconde des 3 classes de maladies histiocytaires (les deux autres étant les histiocytoses langerhansiennes (HCL) (Classe 1) et la classe des histiocytoses malignes[9]). Ces trois classes d'histiocytoses ont en commun d'être « des maladies réactives ou malignes dans lesquelles divers tissus, y compris la peau, sont infiltrés par des cellules de la lignée monocyte-macrophage ». La plupart ont pour origine les dendrocytes dermiques[10].
Le XGJ est la forme la plus fréquente d’histiocytose non langerhansienne et fait partie de celles de ces maladies [11] qui au sein des HCNL sont « cliniquement distinctes » et ont « le même immunophénotype[12] ».
Une hypothèse[9] est que l'histiocytose des dendrocytes dermiques constitue un spectre de maladies où le dendrocyte dermique est présent à un degré progressif de maturation. Sur cet axe, les cellules de l’HCB sont de type "jeunes" (immatures) et associées à des maladies brèves, guérissant spontanément en quelques mois[10].
Description
Ces xanthogranulomes sont généralement asymptomatiques mais peuvent (rarement) s'ulcérer ou saigner[10]..
La première apparition est rapide, et parfois suivie d'autres lésions identiques durant des années[10].
- Aspect externe : ces tumeurs se présentent comme des papules (rondes à ovales) nettement délimitées, lisses
Elles sont fermes (consistance de caoutchouc)[10].
Leur couleur, rose à rouge, est nuancée de jaune et tend à virer au jaune-brun avec le temps[10] ; - Localisation : Les papules apparaissent plus souvent sur la tête et le cou, et moins souvent sur le haut du tronc et les membres[13] et très exceptionnellement sur les muqueuses buccales (souvent après l'âge de 3 ans sur les côtés de la langue ou milieu du palais, pouvant s'ulcérer et saigner)[10], parfois hyperkératosiques ou prenant des formes géantes, ou pédiculées ou même (exceptionnellement) lichénoïdes généralisées[10].
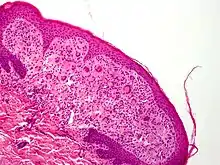
- Aspect histologique : La nature de l'infiltrat diffère selon l'âge de la lésion. L'aspect inflammatoire est moins visible (avec cytoplasme homogène et éosinophilique ou amphophilique) dans les lésions récentes. Ensuite, la part de l’infiltrat inflammatoire augmente et le cytoplasme des histiocytes est vacuolé et xanthomateux. Quand la lésion guérit elle est caractérisée par des fibroblastes et une fibrose. Les cellules géantes sont plus rares dans les lésions précoces et vieilles[14]. Des noyaux atypiques et des figures mitotiques sont parfois visibles [15]. L’activité mitotique est plus visible dans les lésions jeunes précoces[14].
- Taille : variable ; de 5 millimètres de diamètre et de quelques mm d'épaisseur à une tumeur bien visible de plusieurs centimètres de large (jusqu'à 2,5 cm[2] , avec quelques rares cas de forme géante (macronodulaires, jusqu'à 10 cm pouvant être confondues avec un hémangiome type « Cyrano » [16]) ; des cas atypiques existent, avec « éruptions lichénoïdes, réticulées, maculopapuleuses, en plaque, linéaires »[2].
- le xanthogranulome est plus souvent seul (60 % à 82 % des cas[10]), et présent sur la tête (sur le cuir chevelu éventuellement), le cou, tronc et membres.
Si la XGJ touche aussi le tissu sous-cutané (tissu mou profond dans 5 % des cas[10], plus souvent sur tête et au cou[17]), on sent à la palpation une masse nettement limitée, qui peut gêner le patient, la peau en regard pouvant ne pas être modifiée ou devenir jaunâtre[18]. - Des télangiectasies sont parfois (rarement) observées en périphérie ou sur la tumeur [10]. ;
Deux variantes (qui coexistent souvent) ont été définies par Gianotti[19] :
- forme nodulaire petite, avec beaucoup de papules en demi-sphères de 2 à 5 mm de diamètre,
- forme nodulaire de grande taille, avec un seul nodule ou quelques-uns, et plus grands (10 à 20 mm de diamètre.
Prévalence
Elle n'est pas connue dans le monde, elle touche potentiellement toutes les races (peu de cas sont rapportés par la littérature pour les personnes à peau noire)[10]. Chez l'adulte autant de femmes que d'hommes sont touchés, mais chez les jeunes enfants, le sex-ration serait de 1,5 garçons pour 1 fille [10]
Histologie
Les xanthogranulomes contiennent dans un premier temps des histiocytes non lipidiques, sans cellules de langerhans. Ensuite, des cellules inflammatoires (dont des histiocytes spumeux) apparaissent, de plus en plus nombreuses avec la maturation de la tumeur. Des cellules géantes de Touton aux noyaux arrangés en couronne, sont typiques, avec parfois d'autres cellules géantes, « endothéliales »[20].
Histoire
- 1905: un premier cas d'XGJ est rapportée par Adamson[10], dans la littérature anglaise ; c'est celui d'un enfant ayant développé de nombreuses papules de couleur jaune-blanc sur le corps dans les 2 premières semaines de vie. Adamson nomme cette maladie « congenital xanthoma multiplex » (« xanthome disséminé congénital »).
- En 1909 McDonagh étudie un autre cas puis (en 1912 une série de 5 autres cas) ; Considérant cette maladie comme ayant une origine endothéliale, il la nomme « nævo-xantho-endothéliome »[10].
- 1912, McDonagh présente le bilan d'un premier cas rebaptisé « nevoxanthoendothelioma » bien que non associée à des nævus ou des cellules endothéliales).
- En 1936, Senear et Caro montrent que la lésion est en fait xanthomateuse et comme elle affecte les jeunes enfants la nomment « xanthome juvénile »[10].
- 1937 Laurb et Lain rapportent des atteintes viscérales de XGJ ; Ils décrivent un premier cas d'atteinte pulmonaire. D'autres localisations extra-cutanés (confirmés par des biopsies) sont ensuite détectées, dont une première atteinte oculaire (par Blank Eglick et Beerman en 1949), puis (en 1951) sur la muqueuse du palais (« petite papule jaune », et (en 1974 ;premier vrai cas de XGJ intra-buccal) dans la bouche[10].
- 1949 L'équipe de Blank et al. décrit la première atteinte oculaire
- 1954, Helwig et Hackney réintroduisent l'expression « Xanthogranulome juvénile » qui reflète mieux l'aspect histopathologique de la tumeur[10].
Ils ont montré histologiquement (en observant des histiocytes contenant des lipides et des cellules géantes) que ces papules ne proviennent pas de nævi ou de cellules endothéliales[10]. - On constate peu à peu aussi que les enfants sont largement les plus touchés, mais que des adultes le sont aussi parfois[10].
« L'aspect xanthélasmoïde des lésions et les similitudes évolutives entre mastocytose de l'enfant et xanthogranulome juvénile firent suggérer l'hypothèse d'un processus commun avec un spectre histologique allant du mastocytome au xanthome »[21]
Incidence
Son incidence dans les différentes population est inconnue[2].
Selon les données disponibles et pour les pays où elles sont disponibles ;
- 5 à 17 % des nouveau-nés en portent[1].
- 40 à 70 % jeunes enfants (premières années de la vie) en portent aussi[1]
- Les adultes n'en ont que rarement, mais alors ils sont pérennes, apparaissant entre 20 et 30 ans (jusqu'à 80 ans dans quelques cas)[1]
- Caputo et al. en ont observé[16] 104 nouveaux cas en 20 ans dans leurs consultations, dont certains avec des formes atypiques.
Pronostic
Chez l'enfant, la guérison survient spontanément en trois à six ans dans presque tous les cas (selon les données de la littérature, c'est-à-dire pour les cas diagnostiqués) ;
L'adulte ne guérit pas spontanément, ou il reste des séquelles (hyperpigmentation, atrophie, anétodermie)[22],[23].
Des récidives après exérèse ont été observées[13] ainsi que quelques évolutions fatales (toujours en cas de localisations multiples, et en dépit de traitement systémique de type corticothérapie générale à fortes doses, radiothérapie, chimiothérapie, immunosuppresseurs[13],[24]).
Localisation (par ordre de fréquence)
Ce sont le plus souvent les yeux, puis les paupières, devant les poumons (apparence de métastases à la radio) et le foie (avec hépatomégalie souvent). Plus rarement, le péricarde, le myocarde, la rate, le rétropéritoine, les reins, le système nerveux central, les gonades, la glande surrénale, les os et le larynx sont touchés[10]. De manière plus détaillée :
- sur la peau : plutôt tête, cou, tronc et membres, mais potentiellement partout, et sur les muqueuses (bouche, organes génitaux)
- localisation extracutanées :
sur l'œil chez 0,3 à 0,5 % des patients présentant un XGJ[1],[25] selon certains auteurs, plus rare selon d'autres (absence sur des études récentes de 129[15] et 174[24] cas. La lésion est présente dans l’hyphéma souvent, avec au moment du diagnostic œil rouge, douloureux, irrité photophobie [8] et éventuelle hémorragie dans la chambre antérieure, ceci sans atteinte cutanée pour 45 % des patients. Divers autres cas de figures sont possibles (ex : tumeur asymptomatique de l'iris, glaucome unilatéral hyphéma spontané, uvéite, une hétérochromie irienne[1]. Les paupières ou plus rarement le pôle postérieur et de l'orbite peuvent aussi être touchés[1]. Chang et al. ont estimé[25] que le risque d'atteinte intra-oculaire[26] est plus élevé pour les enfants de moins de deux ans présentant des XGJ multiples. Pour eux, Hernandez-Martin et al. recommandent deux consultations ophtalmologiques par an jusqu’à l’âge de deux ans[1]. - localisation pulmonaire, avec dyspnée parfois (opacités arrondies évoquant des métastases à la radiographie) ;
- localisation hépatique, détectée par l'hépatomégalie
- et beaucoup plus rarement (avec sous-estimation possible du nombre de cas en raison d'un diagnostic plus difficile) : rate, pancréas, rétropéritoine, tractus gastro-intestinal, rein, gonades, surrénales, péricarde, myocarde, os, système nerveux central, glandes salivaires et trachée[1],[25],[15],[14],[27]. Ces lésions sont généralement découvertes via les conséquences cliniques d'une compression d'un nerf ou d'un vaisseau par la tumeur. Ces cas sont très souvent associés à des atteintes cutanées multiples, ou à la fois cutanées et sous-cutanées[1],[14].
Diagnostic
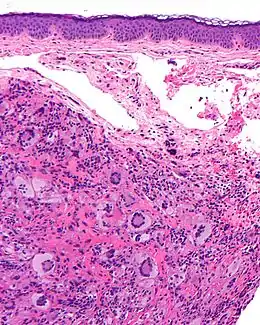
La coupe histologique (grossissement : × 40) montre un infiltrat dermique (dense et bien limité) composé d'histiocytes, de cellules géantes de Touton, et parfois de cellules mastocytaires, et d’un infiltrat inflammatoire lymphocytaire (lymphocytes, éosinophiles, neutrophiles).
L'infiltration affecte le derme papillaire et parfois réticulaire ou le niveau des tissus sous-cutanés, du fascia et des muscles.
Le derme peut être très aminci par l'infiltrat et alors s'ulcérer.
Diagnostic différentiel
Il vise à éliminer les hypothèses d'autres proliférations histiocytaires nodulaires, dont
- histiocytose langerhansienne (histologiquement différente du XGJ)
- histiocytose auto-involutive de Hashimoto-Pritzker (souvent ulcérée et qui guérit en général en quelques mois). En immunohistochimie, le marquage par la PS100 est positif pour 10 à 20 % des cellules.
- histiocytose céphalique bénigne (ou HCB) qui est cependant histologiquement différente du XGJ)[10] mais qu'on ne peut parfois pas différentier histologiquement d’une forme précoce de XGJ. Elle donne cependant des papules plus planes dans l’histiocytose céphalique bénigne et qui sont présentes sur la tête et/ou le cou.
- nævus de Spitz, cliniquement parfois difficile à différentier d'un XGG (⇒ l'examen histologique montre des cellules épithélioïdes fusiformes proliférantes)[10].
- urticaire pigmentaire(UP), car confusion possible avec la forme micronodulaire multiple de XGJ, cette dernière n'activant cependant pas le signe de Darier (démangeaison et urtication provoquées par la friction[10]
- xanthomatose hyperlipémique (plutôt tendineux et périarticulaires[10].
- tumeurs malignes (de type rhabdomyosarcome, fibrosarcome ou fibrohistiocytome malin) en cas de localisation sous-cutanée et profonde. Ici aussi, l'examen histologique confirmera ou non le diagnostic (les atypies nucléaires et mitoses sont très rares et les cellules tumorales absences du XGJ).
Le Xanthogranulome juvénile ne modifie pas les taux de lipides et de cholestérol du plasma sanguin[20].
L'analyse immunohistochimique permet de différentier les XGJ d'autres formes de proliférations histiocytaires ; Les histiocytes sont[2] :
- négatifs pour la PS100 ;
- négatifs le CD1a
- positifs pour la vimentine ;
- positifs le CD68 ;
- positifs le facteur XIIIa.
Associations, facteurs de risque
- La maladie apparait sans anomalies métaboliques comme l'hyperlipémie ou le diabète insipide, qui accompagne parfois d'autres maladies xanthomateuses[13]
- Dans certains cas (antécédents familiaux), il semble y avoir une prédisposition génétique.
- Une association triple avec leucémie et neurofibromatose de type 1 (NF1) est observée (découverte en 1958)[10].
Ceci invite à un suivi particulier des enfants concernés par l'association NF1 + XGJ (bien que sans valeur prédictive à ce jour en raison de la faiblesse statistique des études).. Les leucémies sont alors le plus souvent des leucémies myélomonocytaires juvéniles (LMMJ également dite leucémie myélogène chronique juvénile)
L'association de xanthogranulome(s) et de neurofibromatose NF1 chez un bébé ou jeune enfant doit alerter le médecin sur le risque accru d'apparition de LMMJ (surtout en cas d'antécédents familiaux de NF19[10]. - Une neurofibromatose de type 1 NF1 est parfois associée (18 % des patients porteurs de NF1 présenteraient des XGJ)[12][source insuffisante]. Selon Cambiaghi et al., chez le bébé, des xanthogranulomes associés à plus de 6 taches café au lait de plus de 5 mm de diamètre serait un marqueur de risque de NF1 [13][source insuffisante].
Soins, conduite à tenir
Une lésion unique de ce type, avec un aspect clinique inhabituel doit susciter une biopsie et un examen clinique.
L'examen ophtalmologique est inutile en cas de lésion unique éloignée de l'œil, mais les parents doivent être alertés sur la nécessité de consulter immédiatement en cas de problème oculaires apparaissant chez l'enfant. En cas d'atteinte oculaire, une intervention chirurgicale ou une radiothérapie peuvent être utile. Le moment idéal est encore discuté.
En cas de lésions multiples, Chang et al. recommandent[25] un examen oculaire deux fois par an et jusqu'à l'âge de deux ans. Une NF1 ou d'éventuelles localisations viscérales doivent aussi être recherchées (interrogatoire et recherche d'indices cliniques).
Un XGJ apparaissant chez un enfant porteur d’une NF1 invite à demander une biopsie de confirmation de diagnostic.
Le risque d'éventuelle LMMJC associée invite à une surveillance clinique rapprochée (en recherchant hépatosplénomégalie, adénopathie), avec surveillance sanguine (pour détecter une éventuelle monocytose).
Références
- Hernandez-Martin A, Baselga E, Drolet BA, Esterly NB. Juvenile xanthogranuloma. J Am Acad Dermatol 1997;36:355—67.
- I. Kupfer-Bessaguet, F. Staroz, P. Plantin, Xanthogranulome juvénile Juvenile xanthogranuloma ; la Société française de dermatologie pédiatrique ; Annales de dermatologie et de vénéréologie (2009) 136, 70—73, mis en ligne 2008-12-19 ; doi:10.1016/j.annder.2008.06.014
- Emedicine/Medscape, Glaucoma, Juvenile
- Malbos, S., Guilhou, J. J., Meynadier, J., Barneon, G., & Baldet, P. (1979). Le xanthogranulome de l’adulte. Dermatology, 158(5), 334-342
- Zina, G., & Ressa, P. G. (1969). Xanthogranulome bénin à régression spontanée de l’adulte. Bull Soc Fr Dermatol Syphiligr, 76, 563-565.
- Venencie, P. Y., Doukan, S., Vieillefond, A., Boyer-Neumann C., Toan, N., Beaumont, J., ... & Delfraissy, J.F. (1992). Xanthogranulome nécrobiotique avec paraprotéinémie. In Annales de dermatologie et de vénéréologie (Vol. 119, No. 11, pp. 825-827). Masson (résumé).
- Bara, C., Barbarot, S., Hamidou, M., Cassagnau, E., Renaut, J. J., Triau, S., ... & STALDER, J. F. (2003). Xanthogranulome nécrobiotique révélé par une atteinte péricardique et pulmonaire. In Annales de dermatologie et de vénéréologie (Vol. 130, No. 3, pp. 341-344). Masson (résumé).
- Bourlond, A., Pirard, C., & Eggers, S. (1989) Xanthogranulome nécrobiotique et myélome. Dermatology, 179(3), 139-142.
- Chu AC. The confusing state of the histiocytoses. Br J Dermatol 2002;143:475-476
- Mouza Alsowaidi, Denis Sasseville, Maladies histiocytaires Partie 2 : Histiocytoses non langerhansiennes , Dermatologie / Conférences Scientifiques, mars 2004, Vol. 3, N°3
- James, William D.; Berger, Timothy G.; et al. Andrews' Diseases of the Skin: clinical Dermatology ; Saunders Elsevier ; 2006 ; (ISBN 0-7216-2921-0)
- Mouza Alsowaidi, Denis Sasseville, Maladies histiocytaires Partie 2 : Histiocytoses non langerhansiennes, Dermatologie / Conférences Scientifiques, mars 2004, Vol. 3, N°3
- Hernandez-Martin A, Baselga E, Drolet BA, Esterly NB. Juvenile xanthogranuloma. J Am Acad Dermatol 1997;36:355-367.
- Freyer DR, Kennedy R, Bostrom BC, Kohut G, Dehner LP. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr 1996;129: 227—37
- rapportées dans 10 % de 174 cas pédiatriques étudiés par Dehner LP. in Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol 2003;27:579—93.
- Caputo R, Grimalt R, Gelmetti C, Cottoni F. Unusual aspects of juvenile xanthogranuloma. J Am Acad Dermatol 1993;29:868—70
- Dehner LP. Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol 2003;27:579—93
- Janney CG, Hurt MA, Santa Cruz DJ. Deep juvenile xanthogranuloma. Subcutaneous and intramuscular forms. Am J Surg Pathol 1991;15:150—9.
- Gianotti F, Caputo R, Ermacora E, Gianni E. Benign cephalic histiocytosis. Arch Dermatol 1986;122:1038-1043.
- Iwuagwu FC, Rigby HS, Payne F, Reid CD. Juvenile xanthogranuloma variant: a clinicopathologic case report and review of the literature. Br J Plast Surg 1999;52:591-596.
- Loubeyres, S., Leaute-Labreze, C., Roul, S., Labbe, L., Bioulac-Sage, P., & Taieb, A. (1999) Classification et prise en charge des mastocytoses de l'enfant. In Annales de dermatologie et de vénéréologie (Vol. 126, No. 1, pp. 20-25). Masson (résumé).
- Sannier K, Dompmartin A, Gallet B, Comoz F, Labbé D, Penven K, et al. Involution atrophique de xanthogranulome juvénile. Ann Dermatol Venereol 2003;130: 1047—50.
- Prigent F. Anétodermie secondaire au xanthogranulome juvénile. Ann Dermatol Venereol 2001;128:291.
- Janssen D, Harms D. Juvenile xanthogranuloma in childhood and adolescence. A clinicopathologic study of 129 patients from the kiel pediatric tumor registry. Am J Surg Pathol 2005;29:21—8.
- Chang MW, Frieden IJ, Good W. The risk of intra-ocular juvenile xanthogranuloma: survey of current practices and assessment of risk. J Am Acad Dermatol 1996;34:445—9.
- Hamdani, M., El Kettani, A., Rais, L., El Belhadji, M., Rachid, R., Laouissi, N., ... & Amraoui, A. (2000). Xanthogranulome juvénile avec localisation intra-oculaire: A propos d'un cas. Journal français d'ophtalmologie, 23(8), 817-820.
- Naiman AN, Bouvier R, Colreavy MP, Bellon G, Froehlich P. Tracheal juvenile xanthogranuloma in a child. Int J Pediatr Otorhinolaryngol 2004;68:1469—72.
Voir aussi
Articles connexes
- Xanthogranulome
- Physiopathologie
- Tumeurs de la peau
- Histiocytose non langerhansienne
- Dermatologie
- Tumeur
- Histiocytose
- Histiocytose non langerhansienne
- Macrophages
- Histiocyte
- Dendrocyte
- Xanthome
Liens externes
- (fr) Emedicine/Medscape, llustration (sur le bras d'un bébé)
Bibliographie
- Kupfer-Bessaguet, I., Staroz, F., & Plantin, P. (2009, January). Xanthogranulome juvénile. In Annales de dermatologie et de vénéréologie (Vol. 136, No. 1, p. 70-73). Elsevier Masson.
- Tanz, W. S., Schwartz, R. A., & Janniger, C. K. (1994). Juvenile xanthogranuloma. Cutis, 54(4), 241-245.
- Sannier, K., Dompmartin, A., Gallet, B., Comoz, F., Labbé, D., Penven, K., & Leroy, D. (2003). Involution atrophique de xanthogranulomes juvéniles. In Annales de dermatologie et de vénéréologie (Vol. 130, No. 11, p. 1047-1050). Masson.
ouai chui dakor
 Portail de la médecine
Portail de la médecine