Xeroderma pigmentosum
Maladie des enfants de la Lune
| Xeroderma pigmentosum Syndrome de DeSanctis-CacchioneMaladie des enfants de la Lune | |
| Référence MIM | Voir article |
|---|---|
| Transmission | Récessive |
| Chromosome | Voir article |
| Gène | Voir article |
| Empreinte parentale | Non |
| Anticipation | Non |
| Prévalence | 1 sur 100 000 à 1 000 000 |
| Maladie génétiquement liée | Voir article |
| Diagnostic prénatal | Possible |
| Liste des maladies génétiques à gène identifié | |
| Spécialité | Génétique médicale |
|---|
| CIM-10 | Q82.1 |
|---|---|
| CIM-9 | 757.33 |
| OMIM | 194400 |
| DiseasesDB | 14198 |
| MedlinePlus | 001467 |
| eMedicine | 1177889 |
| MeSH | D014983 |
| Patient UK | Xeroderma-Pigmentosum |
![]() Mise en garde médicale
Mise en garde médicale
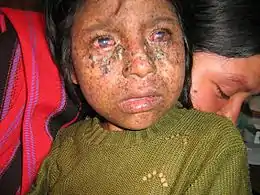
Décrit en 1870[1] par Moritz Kaposi, le xeroderma pigmentosum, ou maladie des « enfants de la Lune », est une maladie héréditaire d'origine génétique très rare (1/1 000 000). Elle se caractérise par une sensibilité excessive de la peau aux rayons ultraviolets, des troubles oculaires et un risque fortement accru de développer un cancer de la peau ou des yeux. Concrètement, les mécanismes de réparation de l'ADN sont inopérants et n'arrivent pas à réparer notamment les dimères de thymine. Plus précisément, ces patients sont déficients dans l'un des gènes codant les protéines participant au mécanisme de réparation par excision de nucléotides. Les mutations dues à l'environnement (surtout les ultraviolets) s'accumulent donc au cours des mitoses successives.
Le xeroderma pigmentosum (XP) touche les deux sexes. En fonction de sa forme et de l'âge auquel il se déclare, il réduit significativement l'espérance de vie du malade.
Il n'existe à l'heure actuelle aucun traitement. On ne peut que limiter les symptômes en appliquant des mesures préventives drastiques et très onéreuses.
Symptômes
La moitié des personnes atteintes ont, dès le plus jeune âge (environ 2 ans), une réaction cutanée excessive à l'exposition solaire. Une des caractéristiques de la maladie est la présence de lentigines solaires semblables à des taches de rousseur importantes au niveau du visage avant l'âge de deux ans, alors que celles-ci sont rares chez les enfants. La peau est sèche et épaisse avec des anomalies variées de la pigmentation.
Les troubles oculaires sont souvent limités à la chambre antérieure de l'œil impliquant une cataracte ou une kératite. Les paupières sont très fragiles avec perte des cils et pouvant aller jusqu'à la destruction totale. L'âge moyen de survenue des cancers de la peau est inférieur à 10 ans.
Un tiers des individus ont des manifestations neurologiques comprenant des pertes de l'audition, une microcéphalie, des troubles du développement et des réflexes tendineux diminués ou absents.
Les formes variantes du xeroderma pigmentosum se manifestent beaucoup plus tard. L'affection se caractérise dès la petite enfance pour la plupart de ses formes par des hyperpigmentations apparaissant au soleil évoluant en cancer ou complications oculaires.
Le xeroderma pigmentosum peut aussi inclure d'autres problèmes neurologiques ou un nanisme.
La personne atteinte doit protéger les fenêtres de sa maison, et du véhicule dans lequel elle circule, par des filtres anti-UV et porter un casque anti-UV.
Causes
La dénomination Xeroderma pigmentosum recouvre un ensemble de syndromes génétiques associés à des mutations dans les différentes protéines impliquées dans la réparation de l’ADN[2]. Plus précisément, les facteurs protéiques mutés participent aux différentes étapes de la réparation par excision de nucléotides, un mécanisme spécifique de correction des lésions encombrantes et/ou étendues sur l'ADN[3].
Chez les mammifères placentaires et en particulier chez l'humain, la réparation par excision de nucléotides est le seul mécanisme de réparation des dimères de pyrimidines, des lésions de l'ADN créées par l'exposition aux UV[3]. La sensibilité particulière des malades atteints de xeroderma pigmentosum à la lumière est une conséquence de l'incapacité de leurs cellules à réparer ces lésions UV-induites.
Aspects génétiques
Le xeroderma pigmentosum est une maladie qui se transmet de génération en génération. L’enfant n’héritera pas de cette maladie si le gène muté n’est présent qu’en un seul exemplaire, ce qui suppose que, pour l’avoir, il faut que les deux parents transmettent le même gène muté en un exemplaire chacun. Contrairement à l’enfant, les parents eux ne sont pas concernés par la maladie puisque porteurs que d’un seul exemplaire. Si les parents ont donné naissance à un enfant malade, la probabilité d’avoir un second enfant atteint de cette même maladie est donc de 25 %.
Cette maladie concerne une à quatre personnes sur 1 000 000. Ce chiffre concerne surtout l’Europe et les États-Unis, on constate en effet une incidence plus grande en Asie et au Moyen-Orient.
Différents types de xeroderma pigmentosum
| Type | fréquence | signes cutanés | clinique | gène | chromosome | M.I.M. |
|---|---|---|---|---|---|---|
| A | 25 % | oui | signes neurologiques | XPA | 9q22.3 | 278700 |
| B | rare | pas obligatoire | syndrome de Cockayne | ERCC3 | 2q21 | 133510 |
| C | 25 % | oui | troubles des ongles et des cheveux | XPC | 3p25 | 278720 |
| D | 15 % | pas obligatoire | troubles des ongles et des cheveux-syndrome de Cockayne-syndrome de Pena-Shokeir-signes neurologiques | ERCC2 | 19q13.2-q13.3 | 278730 |
| E | rare | oui | pas de signes neurologiques | DDB2 | 11p12-p11 | 278740 |
| F | 6 % | oui | pas de signes neurologiques | ERCC4 | 16p13.3-p13.13 | 278760 |
| G | 6 % | oui | syndrome de Cockayne | ERCC5 | 13q33 | 278780 |
| variant | 21 % | oui | pas de signes neurologiques | POLH | 6p21.1-p12 | 200150 |
Diagnostic de la maladie
Généralement la maladie est diagnostiquée assez tôt, en effet des brûlures apparaissent dès les premières expositions au soleil. Ces brûlures très importantes attirent généralement l’attention de l’entourage de l’enfant, ainsi que du médecin. Malgré tout, certaines personnes atteintes ne présentent pas ces brûlures. La maladie n’est donc diagnostiquée qu'à l’apparition des lésions précancéreuses ou cancéreuses sur la peau. Les manifestations au niveau des yeux, en particulier la photophobie, peuvent aussi révéler la maladie. Le xeroderma pigmentosum étant d’origine génétique, le diagnostic est confirmé en analysant l’ADN. Pour cela, on prélève de la peau en profondeur grâce à une biopsie.
Le xeroderma pigmentosum peut parfois être confondu avec d’autres maladies, comme le syndrome de Cockayne ou celui de Bloom. Il peut aussi parfois s’agir d’une simple photosensibilité anormale. L’examen du malade, ainsi que la biopsie de la peau permettent de distinguer les différentes maladies entre elles.
Il est important de dépister cette maladie rapidement, car des mesures protectrices doivent être prises le plus tôt possible.
Évolution et risques
Non ou mal protégée, la peau des sujets atteints peut présenter des lésions photosensibles : dépigmentation, hyperpigmentation, lentigos, télangiectasies, kératoses actiniques et atrophie. Le coefficient multiplicateur concernant le risque de cancer de la peau, hors mélanome, est estimé à 10 000, 2 000 pour le mélanome, avant l'âge de 20 ans[4].
De même, yeux et paupières sont particulièrement sensibles : au moins 40 % des patients présentent blépharospasme et photophobie. Sont également fréquents les érythèmes, troubles de la pigmentation, atrophies et lésions cancéreuses sur les paupières, tout comme les opacifications cornéennes et les ptérygions[4].
Mesures de protection
Le seul moyen de lutter efficacement contre le xeroderma pigmentosum est de protéger la totalité du corps des rayons ultraviolets (UV). De préférence, il faut éviter de sortir le jour et éviter l’exposition à certaines lampes dégageant des UV (lampes dites "néons" ou tubes fluorescents) et mesurer son taux d'UV avant d'y exposer les malades. Sinon lors des sorties, le corps doit être protégé grâce à des vêtements amples, des gants, des chapeaux à large bords et appliquer intensivement et fréquemment des crèmes solaires à indice de protection maximal. Il faut consulter très régulièrement son dermatologue et traiter tout problème dermatologique rapidement.
Il y a quelques années, une combinaison intégrale de protection était développée par la NASA, mais sa production a été arrêtée[Quand ?]. Il n’existe donc pas de système de protection destiné à part entière à cette maladie. Ces mesures sont très coûteuses et très contraignantes, pour le malade qui est souvent coincé chez lui, ce qui implique de grandes difficultés pour la scolarisation et le maintien du lien social, mais aussi pour la famille qui doit s’adapter à cette maladie.
Culture populaire
Dans la littérature
- Chris Snow, le héros de la trilogie de Moonlight Bay du célèbre auteur de best-sellers américain Dean Koontz est un jeune homme atteint de xeroderma pigmentosum.
- Il était une fois sous la lune, de Catherine Lafaye Latteux : une histoire pour les 6-12 ans qui présente la maladie et ses conséquences.
- Mon amoureux de la Lune, de Agnès de Lestrade, sur des illustrations de Amandine Laprun, paru dans Les petites sorcières : une histoire pour les 8-12 ans racontant l'histoire d'une jeune fille qui découvre que son nouveau voisin est atteint de la maladie, mais pourtant, va se lier d'amitié avec lui. Réédité en 2015 chez Oskar.
- Au bout là-bas, de Anne Vantal, chez Actes Sud (9-12 ans) : quel est donc ce mystérieux locataire qui ne sort que la nuit ?
- Deux enfants sous la lune, de Jackie Landreaux, chez J'ai Lu (9-12 ans) : Tim est très intrigué par les enfants de la maison voisine.
- Les enfants de la lune, de Thierry Dedieu, chez Seuil (3-6 ans) : texte poétique pour montrer ce que voient et vivent les enfants qui ne peuvent sortir que la nuit.
- Comte Cain de Kaori Yuki, dans le volume 3 Kafka chapitre 2, Julianne Alche est atteinte de xeroderma pigmentosum. Quand le Comte Cain ouvre les volets, Devon (le frère de Julianne) lui crie de fermer les volets, car le soleil blesse sa sœur, et qu'elle ne peut même pas sortir dehors.
- Susan Hopper, série de Anne Plichota et Cendrine Wolf, édition XO Jeunesse : Susan, orpheline touchée par une malédiction, va se lier d'amitié avec Eliot, le fils de ses parents adoptifs, atteint de Xeroderma Pigmentosum.
- Dans Le survivant de Christian Lamblin, l'un des personnages principaux, Tom, est atteint du xeroderma pigmentosum et porte une combinaison spéciale pour le protéger du soleil durant le jour.
- Dans Journal d'un enfant de lune, bande dessinée de Joris Chamblain et Anne-Lise Nalin, Morgane découvre dans sa nouvelle chambre le journal de Maxime, un enfant de la lune.
Dans la cinématographie
- Dans la série japonaise et le film japonais de même nom : Taiyō no Uta, l'héroïne, incarnée respectivement par Erika Sawajiri et par YUI, est atteinte de la maladie.
- Alejandro Amenábar, dans le film Les Autres, avec Nicole Kidman, s'est inspiré du xeroderma pigmentosum pour la maladie dont les deux enfants sont atteints. Ils vivent cloîtrés dans un manoir, mais il n'est pas clairement dit qu'il s'agisse du xeroderma pigmentosum. Dans les bonus du film, il y a par ailleurs un reportage sur la famille Séris, fondatrice de l'association Enfants de la Lune, et dont deux enfants sont atteints de la maladie.
- En 2011, Delphine Gleize fait de cette maladie le sujet central de son film La Permission de minuit.
- En 2011, Jane Gull en fait également un court-métrage Sunny Boy.
- En 2018, Scott Speer qui présente le film Midnight Sun. C'est un film tragique - une jeune fille nommée Katie atteinte de la maladie Xeroderma pigmentosum tombe amoureuse de Charlie le nageur.
Dans la musique
- Le groupe britannique Alan Parson Project [5] a, dans Eye in the Sky (album) parlé de cette maladie rare dans le titre "Children of the Moon"
- Le rappeur français Gringe a sorti le son premier album, intitulé "Enfant Lune"
Notes et références
- (en) Hebra F. et Kaposi M « On disease of the skin, including exanthemata » London - New Sydenham Society.
- C. Robert, A. Sarasin, « Xeroderma pigmentosum », sur www.therapeutique-dermatologique.org, (consulté le )
- Geoffrey M. Cooper (trad. de l'anglais), La cellule : une approche moléculaire, Paris, Bruxelles, de Boeck, , 674 p. (ISBN 2-7445-0056-9, lire en ligne), pp190-196
- (en) Ramkumar HL, Brooks BP, Cao X, Tamura D, Digiovanna JJ, Kraemer KH, Chan CC, « Ophthalmic manifestations and histopathology of xeroderma pigmentosum: two clinicopathological cases and a review of the literature », Surv Ophthalmol, vol. 56, no 4, , p. 348-61. (PMID 21684361, PMCID PMC3137889, DOI 10.1016/j.survophthal.2011.03.001)
- Alan Parsons Project., The best of the Alan Parson Project, Wise, (ISBN 0-7119-0339-5 et 9780711903395, OCLC 63796310, lire en ligne)
Voir aussi
Articles connexes
maladie génétique | épiderme | ultraviolet | mélanine | mitose | cancer | Acide désoxyribonucléique | Syndrome de Cockayne
Liens externes
- Page spécifique sur Orphanet
- (en) Daniel J Wattendorf, Kenneth H Kraemer, « Xeroderma Pigmentosum » in GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2006
- (it)(en) Site
 Portail de la médecine
Portail de la médecine