Galactosemia (British Galactosaemia) is a rare genetic metabolic disorder that affects an individual's ability to metabolize the sugar galactose properly. Although the sugar and lactose metabolizes to galactose, galactosemia is not related to and should not be confused with lactose intolerance. Galactosemia follows an autosomal recessive mode of inheritance that confers a deficiency in an enzyme responsible for adequate galactose degradation. Its incidence is about one per 60,000 births for Caucasians. In other populations the incidence rate differs.
Lactose in food (such as dairy products) is broken down by the enzyme lactase into glucose and galactose. In individuals with galactosemia, the enzymes needed for further metabolism of galactose are severely diminished or missing entirely, leading to toxic levels of galactose 1-phosphate in various tissues as in the case of classic galactosemia, resulting in hepatomegaly (an enlarged liver), cirrhosis, renal failure, cataracts, brain damage, and ovarian failure. Without treatment, mortality in infants with galactosemia is about 75%.
Diagnosis
Infants are routinely screened for galactosemia in the United States. Infants affected by galactosemia typically present with symptoms of lethargy, vomiting, diarrhea, failure to thrive, and jaundice. If the family of the baby has a history of galactosemia, doctors can test prior to birth by taking a sample of fluid from around the fetus or from the placenta.
Detection of the disorder through newborn screening (NBS) does not depend on protein or lactose ingestion, and, therefore, it should be identified on the first specimen unless the infant has been transfused. A specimen should be taken prior to transfusion. The enzyme is prone to damage if analysis of the sample is delayed or exposed to high temperatures.
Treatment
The only treatment for classic galactosemia is eliminating lactose and galactose from the diet. Even with an early diagnosis and a restricted diet, however, some individuals with galactosemia experience long-term complications such as speech difficulties, learning disabilities, neurological impairment (e.g. tremors, etc. ), and ovarian failure in females. Infants with classic galactosemia cannot be breast-fed due to lactose in human breast milk and are usually fed a soy-based formula.
Galactosemia is sometimes confused with lactose intolerance, but galactosemia is a more serious condition. Lactose intolerant individuals have an acquired or inherited shortage of the enzyme lactase, and experience abdominal pains after ingesting dairy products, but no long-term effects. In contrast, a galactosemic individual who consumes galactose can cause permanent damage to their bodies.
Glycogen Storage Disease
Glycogen storage disease (GSD, also glycogenosis and dextrinosis) is the result of defects in the processing of glycogen synthesis or breakdown within muscles, liver, and other cell types. GSD has two classes of cause: genetic and acquired. Genetic GSD is caused by any inborn error of metabolism (genetically defective enzymes) involved in these processes. In livestock, acquired GSD is caused by intoxication with the alkaloid castanospermine. Overall, according to a study in British Columbia, approximately 2.3 children per 100,000 births (one in 43,000) have some form of glycogen storage disease. In the United States, they are estimated to occur in one per 20,000-25,000 births. A Dutch study estimated it to be one in 40,000.
There are 11 distinct diseases that are commonly considered to be glycogen storage diseases (some previously thought to be distinct have been reclassified).
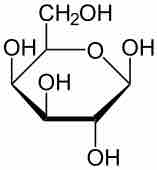
Galactose
Galactosemia is caused by the inability to metabolize galactose, shown here.