Identification of bacteria in the laboratory is particularly relevant in medicine, where the correct treatment is determined by the bacterial species causing an infection. Consequently, the need to identify human pathogens was a major impetus for the development of techniques to identify bacteria.
Early studies have shown that the microbial life around us in the air, sea, and soil is very diverse and only a small fraction of the species are known. One limitation of identifying human pathogens or conventional sequencing begins with a culture of identical cells as a source of DNA. However, early metagenomic studies revealed that there are probably large groups of microorganisms in many environments that cannot be cultured and thus cannot be sequenced. These early studies focused on 16S ribosomal RNA sequences which are relatively short, often conserved within a species, and generally different between species. Many 16S rRNA sequences have been found which do not belong to any known cultured species, indicating that there are numerous non-isolated organisms out there. These surveys of ribosomal RNA (rRNA) genes taken directly from the environment revealed that cultivation based methods find less than 1% of the bacterial and archaeal species in a sample.
The discovery of such diversity led to the field of metagenomics, which is the study of metagenomes, genetic material recovered directly from environmental samples. Rather than culturing a microbe, this approach takes a sample and identifies the different species in it by sequencing all the species simultaneously. However, recovery of DNA sequences longer than a few thousand base pairs from environmental samples was very difficult until recent advances in molecular biological techniques. More specifically, the construction of libraries in bacterial artificial chromosomes (BACs) provided better vectors for molecular cloning.
Advances in bioinformatics, refinements of DNA amplification, and the proliferation of computational power have greatly aided the analysis of DNA sequences recovered from environmental samples. These advances have allowed the adaptation of shotgun sequencing to metagenomic samples . The approach, used to sequence many cultured microorganisms and the human genome, randomly shears DNA, sequences many short sequences, and reconstructs them into a consensus sequence.
Shotgun sequencing and screens of clone libraries reveal genes present in environmental samples. This can be helpful in understanding the ecology of a community, particularly if multiple samples are compared to each other. This was further followed by high-throughput sequencing which did the same process as the shotgun sequencing but at a much bigger scale in terms of the amount of DNA that could sequenced from one sample. This provides information both on which organisms are present and what metabolic processes are possible in the community. Using metagenomics, and the resultant sequencing of uncultured microbes, metagenomics has the potential to advance knowledge in a wide variety of fields. It can also be applied to solve practical challenges in medicine, engineering, agriculture, and sustainability.
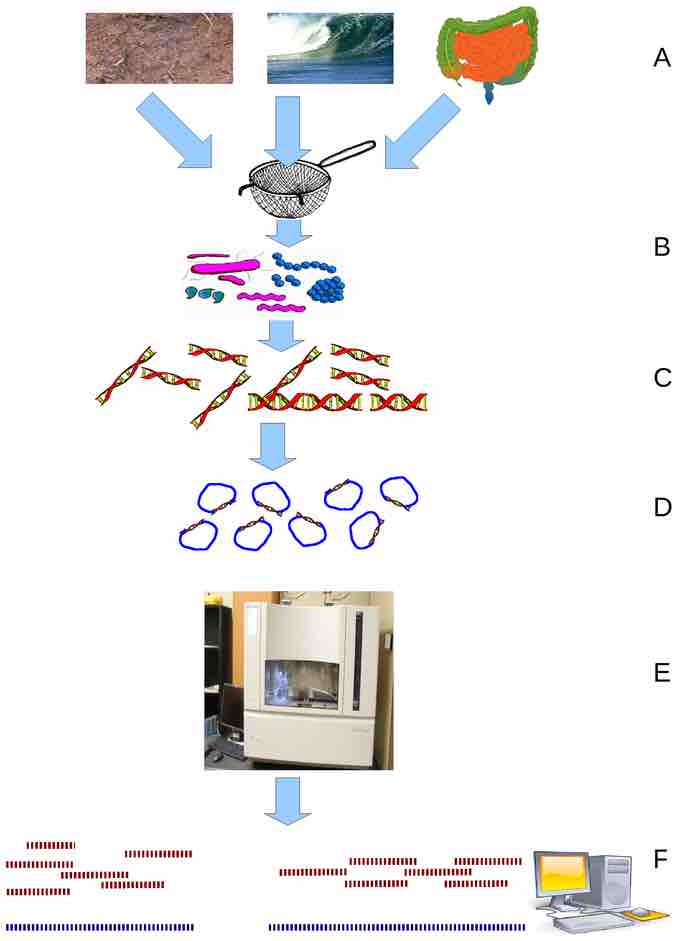
Environmental Shotgun Sequencing (ESS)
(A) sampling from habitat; (B) filtering particles, typically by size; (C) Lysis and DNA extraction; (D) cloning and library construction; (E) sequencing the clones; (F) sequence assembly into contigs and scaffolds