Adaptive immune system
The adaptive immune system, also known as the acquired immune system, is a subsystem of the immune system that is composed of specialized, systemic cells and processes that eliminate pathogens or prevent their growth. The acquired immune system is one of the two main immunity strategies found in vertebrates (the other being the innate immune system).
Like the innate system, the adaptive immune system includes both humoral immunity components and cell-mediated immunity components and destroys invading pathogens. Unlike the innate immune system, which is pre-programmed to react to common broad categories of pathogen, the adaptive immune system is highly specific to each particular pathogen the body has encountered.
Adaptive immunity creates immunological memory after an initial response to a specific pathogen, and leads to an enhanced response to future encounters with that pathogen. Antibodies are a critical part of the adaptive immune system. Adaptive immunity can provide long-lasting protection, sometimes for the person's entire lifetime. For example, someone who recovers from measles is now protected against measles for their lifetime; in other cases it does not provide lifetime protection, as with chickenpox. This process of adaptive immunity is the basis of vaccination.
The cells that carry out the adaptive immune response are white blood cells known as lymphocytes. B cells and T cells, two different types of lymphocytes, carry out the main activities: antibody responses, and cell-mediated immune response. In antibody responses, B cells are activated to secrete antibodies, which are proteins also known as immunoglobulins. Antibodies travel through the bloodstream and bind to the foreign antigen causing it to inactivate, which does not allow the antigen to bind to the host.[1] Antigens are any substances that elicit the adaptive immune response. Sometimes the adaptive system is unable to distinguish harmful from harmless foreign molecules; the effects of this may be hayfever, asthma, or any other allergy.
In adaptive immunity, pathogen-specific receptors are "acquired" during the lifetime of the organism (whereas in innate immunity pathogen-specific receptors are already encoded in the genome). This acquired response is called "adaptive" because it prepares the body's immune system for future challenges (though it can actually also be maladaptive when it results in allergies or autoimmunity).
The system is highly adaptable because of two factors. First, somatic hypermutation is a process of accelerated random genetic mutations in the antibody-coding genes, which allows antibodies with novel specificity to be created. Second, V(D)J recombination randomly selects one variable (V), one diversity (D), and one joining (J) region for genetic recombination and discards the rest, which produces a highly unique combination of antigen-receptor gene segments in each lymphocyte. This mechanism allows a small number of genetic segments to generate a vast number of different antigen receptors, which are then uniquely expressed on each individual lymphocyte. Since the gene rearrangement leads to an irreversible change in the DNA of each cell, all progeny (offspring) of that cell inherit genes that encode the same receptor specificity, including the memory B cells and memory T cells that are the keys to long-lived specific immunity.
Naming
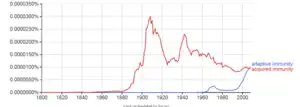
The term "adaptive" was first used by Robert Good in reference to antibody responses in frogs as a synonym for "acquired immune response" in 1964. Good acknowledged he used the terms as synonyms but explained only that he preferred to use the term "adaptive". He might have been thinking of the then not implausible theory of antibody formation in which antibodies were plastic and could adapt themselves to the molecular shape of antigens, and/or to the concept of "adaptive enzymes" as described by Monod in bacteria, that is, enzymes whose expression could be induced by their substrates. The phrase was used almost exclusively by Good and his students and a few other immunologists working with marginal organisms until the 1990s when it became widely used in tandem with the term "innate immunity" which became a popular subject after the discovery of the Toll receptor system in Drosophila, a previously marginal organism for the study of immunology. The term "adaptive" as used in immunology is problematic as acquired immune responses can be both adaptive and maladaptive in the physiological sense. Indeed, both acquired and innate immune responses can be both adaptive and maladaptive in the evolutionary sense. Most textbooks today, following the early use by Janeway, use "adaptive" almost exclusively and noting in glossaries that the term is synonymous with "acquired".
The classic sense of "acquired immunity" came to mean, since Tonegawa's discovery, "antigen-specific immunity mediated by somatic gene rearrangements that create clone-defining antigen receptors". In the last decade, the term "adaptive" has been increasingly applied to another class of immune response not so-far associated with somatic gene rearrangements. These include expansion of natural killer (NK) cells with so-far unexplained specificity for antigens, expansion of NK cells expressing germ-line encoded receptors, and activation of other innate immune cells to an activated state that confers a short-term "immune memory". In this sense, "adaptive immunity" more closely resembles the concept of "activated state" or "heterostasis", thus returning in sense to the physiological sense of "adaptation" to environmental changes.
Functions
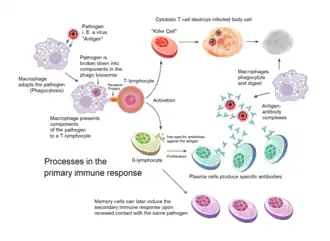
Acquired immunity is triggered in vertebrates when a pathogen evades the innate immune system and (1) generates a threshold level of antigen and (2) generates "stranger" or "danger" signals activating dendritic cells.[2]
The major functions of the acquired immune system include:
- Recognition of specific "non-self" antigens in the presence of "self", during the process of antigen presentation.
- Generation of responses that are tailored to maximally eliminate specific pathogens or pathogen-infected cells.
- Development of immunological memory, in which pathogens are "remembered" through memory B cells and memory T cells.
In humans, it takes 4-7 days for the adaptive immune system to mount a significant response.[3]
Lymphocytes
T and B lymphocytes are the cells of the adaptive immune system. The human body has about 2 trillion lymphocytes, which are 20–40% of white blood cells; their total mass is about the same as the brain or liver. The peripheral bloodstream contains only 2% of all circulating lymphocytes; the other 98% move within tissues and the lymphatic system, which includes the lymph nodes and spleen.[1] In humans, approximately 1–2% of the lymphocyte pool recirculates each hour to increase the opportunity for the cells to encounter the specific pathogen and antigen that they react to.[4]
B cells and T cells are derived from the same multipotent hematopoietic stem cells, and look identical to one another until after they are activated. B cells play a large role in the humoral immune response, whereas T cells are intimately involved in cell-mediated immune responses. In all vertebrates except Agnatha, B cells and T cells are produced by stem cells in the bone marrow.[5] T cell progenitors then migrate from the bone marrow to the thymus, where they develop further.
In an adult animal, the peripheral lymphoid organs contain a mixture of B and T cells in at least three stages of differentiation:
- Naive B and naive T cells, which have left the bone marrow or thymus and entered the lymphatic system, but have yet to encounter their matching antigen
- Effector cells that have been activated by their matching antigen, and are actively involved in eliminating a pathogen
- Memory cells, the survivors of past infections
Antigen presentation
Acquired immunity relies on the capacity of immune cells to distinguish between the body's own cells and unwanted invaders. The host's cells express "self" antigens. These antigens are different from those on the surface of bacteria or on the surface of virus-infected host cells ("non-self" or "foreign" antigens). The acquired immune response is triggered by recognizing foreign antigen in the cellular context of an activated dendritic cell.
With the exception of non-nucleated cells (including erythrocytes), all cells are capable of presenting antigen through the function of major histocompatibility complex (MHC) molecules.[5] Some cells are specially equipped to present antigen, and to prime naive T cells. Dendritic cells, B-cells, and macrophages are equipped with special "co-stimulatory" ligands recognized by co-stimulatory receptors on T cells, and are termed professional antigen-presenting cells (APCs).
Several T cells subgroups can be activated by professional APCs, and each type of T cell is specially equipped to deal with each unique toxin or microbial pathogen. The type of T cell activated, and the type of response generated, depends, in part, on the context in which the APC first encountered the antigen.[2]
Exogenous antigens
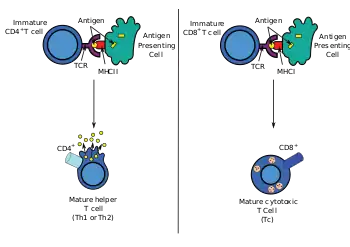
Dendritic cells engulf exogenous pathogens, such as bacteria, parasites or toxins in the tissues and then migrate, via chemotactic signals, to the T cell-enriched lymph nodes. During migration, dendritic cells undergo a process of maturation in which they lose most of their ability to engulf other pathogens, and develop an ability to communicate with T-cells. The dendritic cell uses enzymes to chop the pathogen into smaller pieces, called antigens. In the lymph node, the dendritic cell displays these non-self antigens on its surface by coupling them to a receptor called the major histocompatibility complex, or MHC (also known in humans as human leukocyte antigen (HLA)). This MHC-antigen complex is recognized by T-cells passing through the lymph node. Exogenous antigens are usually displayed on MHC class II molecules, which activate CD4+T helper cells.[2]
Endogenous antigens
Endogenous antigens are produced by intracellular bacteria and viruses replicating within a host cell. The host cell uses enzymes to digest virally associated proteins and displays these pieces on its surface to T-cells by coupling them to MHC. Endogenous antigens are typically displayed on MHC class I molecules, and activate CD8+ cytotoxic T-cells. With the exception of non-nucleated cells (including erythrocytes), MHC class I is expressed by all host cells.[2]
T lymphocytes
CD8+ T lymphocytes and cytotoxicity
Cytotoxic T cells (also known as TC, killer T cell, or cytotoxic T-lymphocyte (CTL)) are a sub-group of T cells that induce the death of cells that are infected with viruses (and other pathogens), or are otherwise damaged or dysfunctional.[2]
Naive cytotoxic T cells are activated when their T-cell receptor (TCR) strongly interacts with a peptide-bound MHC class I molecule. This affinity depends on the type and orientation of the antigen/MHC complex, and is what keeps the CTL and infected cell bound together.[2] Once activated, the CTL undergoes a process called clonal selection, in which it gains functions and divides rapidly to produce an army of “armed” effector cells. Activated CTL then travels throughout the body searching for cells that bear that unique MHC Class I + peptide.
When exposed to these infected or dysfunctional somatic cells, effector CTL release perforin and granulysin: cytotoxins that form pores in the target cell's plasma membrane, allowing ions and water to flow into the infected cell, and causing it to burst or lyse. CTL release granzyme, a serine protease encapsulated in a granule that enters cells via pores to induce apoptosis (cell death). To limit extensive tissue damage during an infection, CTL activation is tightly controlled and in general requires a very strong MHC/antigen activation signal, or additional activation signals provided by "helper" T-cells (see below).[2]
On resolution of the infection, most effector cells die and phagocytes clear them away—but a few of these cells remain as memory cells.[5] On a later encounter with the same antigen, these memory cells quickly differentiate into effector cells, dramatically shortening the time required to mount an effective response.
Helper T-cells
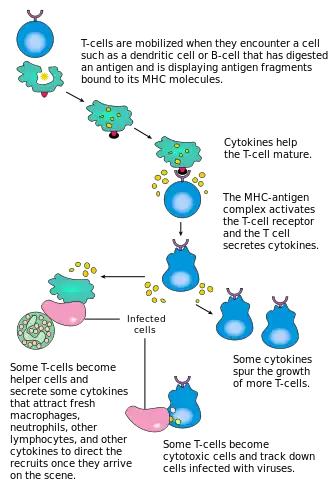
CD4+ lymphocytes, also called "helper" T cells, are immune response mediators, and play an important role in establishing and maximizing the capabilities of the acquired immune response.[2] These cells have no cytotoxic or phagocytic activity; and cannot kill infected cells or clear pathogens, but, in essence "manage" the immune response, by directing other cells to perform these tasks.
Helper T cells express T cell receptors (TCR) that recognize antigen bound to Class II MHC molecules. The activation of a naive helper T-cell causes it to release cytokines, which influences the activity of many cell types, including the APC (Antigen-Presenting Cell) that activated it. Helper T-cells require a much milder activation stimulus than cytotoxic T cells. Helper T cells can provide extra signals that "help" activate cytotoxic cells.[5]
Th1 and Th2: helper T cell responses
Classically, two types of effector CD4+ T helper cell responses can be induced by a professional APC, designated Th1 and Th2, each designed to eliminate different types of pathogens. The factors that dictate whether an infection triggers a Th1 or Th2 type response are not fully understood, but the response generated does play an important role in the clearance of different pathogens.[2]
The Th1 response is characterized by the production of Interferon-gamma, which activates the bactericidal activities of macrophages, and induces B cells to make opsonizing (marking for phagocytosis) and complement-fixing antibodies, and leads to cell-mediated immunity.[2] In general, Th1 responses are more effective against intracellular pathogens (viruses and bacteria that are inside host cells).
The Th2 response is characterized by the release of Interleukin 5, which induces eosinophils in the clearance of parasites.[7] Th2 also produce Interleukin 4, which facilitates B cell isotype switching.[2] In general, Th2 responses are more effective against extracellular bacteria, parasites including helminths and toxins.[2] Like cytotoxic T cells, most of the CD4+ helper cells die on resolution of infection, with a few remaining as CD4+ memory cells.
Increasingly, there is strong evidence from mouse and human-based scientific studies of a broader diversity in CD4+ effector T helper cell subsets.[8][9] Regulatory T (Treg) cells, have been identified as important negative regulators of adaptive immunity as they limit and suppress the immune system to control aberrant immune responses to self-antigens; an important mechanism in controlling the development of autoimmune diseases.[5] Follicular helper T (Tfh) cells are another distinct population of effector CD4+ T cells that develop from naive T cells post-antigen activation. Tfh cells are specialized in helping B cell humoral immunity as they are uniquely capable of migrating to follicular B cells in secondary lymphoid organs and provide them positive paracrine signals to enable the generation and recall production of high-quality affinity-matured antibodies. Similar to Tregs, Tfh cells also play a role in immunological tolerance as an abnormal expansion of Tfh cell numbers can lead to unrestricted autoreactive antibody production causing severe systemic autoimmune disorders.[10]
The relevance of CD4+ T helper cells is highlighted during an HIV infection. HIV is able to subvert the immune system by specifically attacking the CD4+ T cells, precisely the cells that could drive the clearance of the virus, but also the cells that drive immunity against all other pathogens encountered during an organism's lifetime.[5]
Gamma delta T cells
Gamma delta T cells (γδ T cells) possess an alternative T cell receptor (TCR) as opposed to CD4+ and CD8+ αβ T cells and share characteristics of helper T cells, cytotoxic T cells and natural killer cells. Like other 'unconventional' T cell subsets bearing invariant TCRs, such as CD1d-restricted natural killer T cells, γδ T cells exhibit characteristics that place them at the border between innate and acquired immunity. On one hand, γδ T cells may be considered a component of adaptive immunity in that they rearrange TCR genes via V(D)J recombination, which also produces junctional diversity, and develop a memory phenotype. On the other hand, however, the various subsets may also be considered part of the innate immune system where a restricted TCR or NK receptors may be used as a pattern recognition receptor. For example, according to this paradigm, large numbers of Vγ9/Vδ2 T cells respond within hours to common molecules produced by microbes, and highly restricted intraepithelial Vδ1 T cells respond to stressed epithelial cells.
B lymphocytes and antibody production
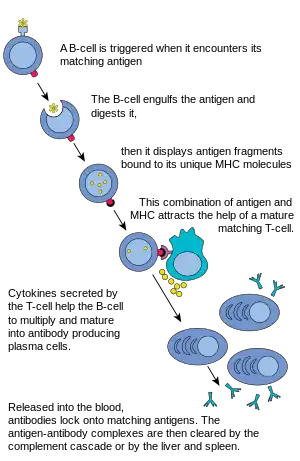
B Cells are the major cells involved in the creation of antibodies that circulate in blood plasma and lymph, known as humoral immunity. Antibodies (also known as immunoglobulin, Ig), are large Y-shaped proteins used by the immune system to identify and neutralize foreign objects. In mammals, there are five types of antibody: IgA, IgD, IgE, IgG, and IgM, differing in biological properties; each has evolved to handle different kinds of antigens. Upon activation, B cells produce antibodies, each of which recognize a unique antigen, and neutralizing specific pathogens.[2]
Antigen and antibody binding would cause five different protective mechanisms:
- Agglutination: Reduces number of infectious units to be dealt with
- Activation of complement: Cause inflammation and cell lysis
- Opsonization: Coating antigen with antibody enhances phagocytosis
- Antibody-dependent cell-mediated cytotoxicity: Antibodies attached to target cell cause destruction by macrophages, eosinophils, and NK cells
- Neutralization: Blocks adhesion of bacteria and viruses to mucosa
Like the T cell, B cells express a unique B cell receptor (BCR), in this case, a membrane-bound antibody molecule. All the BCR of any one clone of B cells recognizes and binds to only one particular antigen. A critical difference between B cells and T cells is how each cell "sees" an antigen. T cells recognize their cognate antigen in a processed form – as a peptide in the context of an MHC molecule,[2] whereas B cells recognize antigens in their native form.[2] Once a B cell encounters its cognate (or specific) antigen (and receives additional signals from a helper T cell (predominately Th2 type)), it further differentiates into an effector cell, known as a plasma cell.[2]
Plasma cells are short-lived cells (2–3 days) that secrete antibodies. These antibodies bind to antigens, making them easier targets for phagocytes, and trigger the complement cascade.[2] About 10% of plasma cells survive to become long-lived antigen-specific memory B cells.[2] Already primed to produce specific antibodies, these cells can be called upon to respond quickly if the same pathogen re-infects the host, while the host experiences few, if any, symptoms.
Alternative systems
In jawless vertebrates
Primitive jawless vertebrates, such as the lamprey and hagfish, have an adaptive immune system that shows 3 different cell lineages, each sharing a common origin with B cells, αβ T cells, and innate-like γΔ T cells.[11][12] Instead of the classical antibodies and T cell receptors, these animals possess a large array of molecules called variable lymphocyte receptors (VLRs for short) that, like the antigen receptors of jawed vertebrates, are produced from only a small number (one or two) of genes. These molecules are believed to bind pathogenic antigens in a similar way to antibodies, and with the same degree of specificity.[13]
In insects
For a long time it was thought that insects and other invertebrates possess only innate immune system. However, in recent years some of the basic hallmarks of adaptive immunity have been discovered in insects. Those traits are immune memory and specificity. Although the hallmarks are present the mechanisms are different from those in vertebrates.
Immune memory in insects was discovered through the phenomenon of priming. When insects are exposed to non-lethal dose or heat killed bacteria they are able to develop a memory of that infection that allows them to withstand otherwise lethal dose of the same bacteria they were exposed to before.[14][15] Unlike in vertebrates, insects do not possess cells specific for adaptive immunity. Instead those mechanisms are mediated by hemocytes. Hemocytes function similarly to phagocytes and after priming they are able to more effectively recognize and engulf the pathogen.[16] It was also shown that it is possible to transfer the memory into offspring. For example, in honeybees if the queen is infected with bacteria then the newly born workers have enhanced abilities in fighting with the same bacteria.[17] Other experimental model based on red flour beetle also showed pathogen specific primed memory transfer into offspring from both mothers and fathers.[18]
Most commonly accepted theory of the specificity is based on Dscam gene. Dscam gene also known as Down syndrome cell adhesive molecule is a gene that contains 3 variable Ig domains. Those domains can be alternatively spliced reaching high numbers of variations.[19] It was shown that after exposure to different pathogens there are different splice forms of dscam produced. After the animals with different splice forms are exposed to the same pathogen only the individuals with the splice form specific for that pathogen survive.[19]
Other mechanisms supporting the specificity of insect immunity is RNA interference (RNAi). RNAi is a form of antiviral immunity with high specificity.[20] It has several different pathways that all end with the virus being unable to replicate. One of the pathways is siRNA in which long double stranded RNA is cut into pieces that serve as templates for protein complex Ago2-RISC that finds and degrades complementary RNA of the virus. MiRNA pathway in cytoplasm binds to Ago1-RISC complex and functions as a template for viral RNA degradation. Last one is piRNA where small RNA binds to the Piwi protein family and controls transposones and other mobile elements.[21] Despite the research the exact mechanisms responsible for immune priming and specificity in insects are not well described.
Immunological memory
When B cells and T cells are activated some become memory B cells and some memory T cells. Throughout the lifetime of an animal these memory cells form a database of effective B and T lymphocytes. Upon interaction with a previously encountered antigen, the appropriate memory cells are selected and activated. In this manner, the second and subsequent exposures to an antigen produce a stronger and faster immune response. This is "adaptive" in the sense that the body's immune system prepares itself for future challenges, but is "maladaptive" of course if the receptors are autoimmune. Immunological memory can be in the form of either passive short-term memory or active long-term memory.
Passive memory
Passive memory is usually short-term, lasting between a few days and several months. Newborn infants have had no prior exposure to microbes and are particularly vulnerable to infection. Several layers of passive protection are provided by the mother. In utero, maternal IgG is transported directly across the placenta, so that, at birth, human babies have high levels of antibodies, with the same range of antigen specificities as their mother.[2] Breast milk contains antibodies (mainly IgA) that are transferred to the gut of the infant, protecting against bacterial infections, until the newborn can synthesize its own antibodies.[2]
This is passive immunity because the fetus does not actually make any memory cells or antibodies: It only borrows them. Short-term passive immunity can also be transferred artificially from one individual to another via antibody-rich serum.
Active memory
In general, active immunity is long-term and can be acquired by infection followed by B cell and T cell activation, or artificially acquired by vaccines, in a process called immunization.
Immunization
Historically, infectious disease has been the leading cause of death in the human population. Over the last century, two important factors have been developed to combat their spread: sanitation and immunization.[5] Immunization (commonly referred to as vaccination) is the deliberate induction of an immune response, and represents the single most effective manipulation of the immune system that scientists have developed.[5] Immunizations are successful because they utilize the immune system's natural specificity as well as its inducibility.
The principle behind immunization is to introduce an antigen, derived from a disease-causing organism, that stimulates the immune system to develop protective immunity against that organism, but that does not itself cause the pathogenic effects of that organism. An antigen (short for antibody generator), is defined as any substance that binds to a specific antibody and elicits an adaptive immune response.[1]
Most viral vaccines are based on live attenuated viruses, whereas many bacterial vaccines are based on acellular components of microorganisms, including harmless toxin components.[1] Many antigens derived from acellular vaccines do not strongly induce an adaptive response, and most bacterial vaccines require the addition of adjuvants that activate the antigen-presenting cells of the innate immune system to enhance immunogenicity.[5]
Immunological diversity

Most large molecules, including virtually all proteins and many polysaccharides, can serve as antigens.[2] The parts of an antigen that interact with an antibody molecule or a lymphocyte receptor, are called epitopes, or antigenic determinants. Most antigens contain a variety of epitopes and can stimulate the production of antibodies, specific T cell responses, or both.[2] A very small proportion (less than 0.01%) of the total lymphocytes are able to bind to a particular antigen, which suggests that only a few cells respond to each antigen.[5]
For the acquired response to "remember" and eliminate a large number of pathogens the immune system must be able to distinguish between many different antigens,[1] and the receptors that recognize antigens must be produced in a huge variety of configurations, in essence one receptor (at least) for each different pathogen that might ever be encountered. Even in the absence of antigen stimulation, a human can produce more than 1 trillion different antibody molecules.[5] Millions of genes would be required to store the genetic information that produces these receptors, but, the entire human genome contains fewer than 25,000 genes.[22]
Myriad receptors are produced through a process known as clonal selection.[1][2] According to the clonal selection theory, at birth, an animal randomly generates a vast diversity of lymphocytes (each bearing a unique antigen receptor) from information encoded in a small family of genes. To generate each unique antigen receptor, these genes have undergone a process called V(D)J recombination, or combinatorial diversification, in which one gene segment recombines with other gene segments to form a single unique gene. This assembly process generates the enormous diversity of receptors and antibodies, before the body ever encounters antigens, and enables the immune system to respond to an almost unlimited diversity of antigens.[2] Throughout an animal's lifetime, lymphocytes that can react against the antigens an animal actually encounters are selected for action—directed against anything that expresses that antigen.
Note that the innate and acquired portions of the immune system work together, not in spite of each other. The acquired arm, B, and T cells couldn't function without the innate system input. T cells are useless without antigen-presenting cells to activate them, and B cells are crippled without T cell help. On the other hand, the innate system would likely be overrun with pathogens without the specialized action of the adaptive immune response.
Acquired immunity during pregnancy
The cornerstone of the immune system is the recognition of "self" versus "non-self". Therefore, the mechanisms that protect the human fetus (which is considered "non-self") from attack by the immune system, are particularly interesting. Although no comprehensive explanation has emerged to explain this mysterious, and often repeated, lack of rejection, two classical reasons may explain how the fetus is tolerated. The first is that the fetus occupies a portion of the body protected by a non-immunological barrier, the uterus, which the immune system does not routinely patrol.[2] The second is that the fetus itself may promote local immunosuppression in the mother, perhaps by a process of active nutrient depletion.[2] A more modern explanation for this induction of tolerance is that specific glycoproteins expressed in the uterus during pregnancy suppress the uterine immune response (see eu-FEDS).
During pregnancy in viviparous mammals (all mammals except Monotremes), endogenous retroviruses (ERVs) are activated and produced in high quantities during the implantation of the embryo. They are currently known to possess immunosuppressive properties, suggesting a role in protecting the embryo from its mother's immune system. Also, viral fusion proteins cause the formation of the placental syncytium[23] to limit exchange of migratory cells between the developing embryo and the body of the mother (something an epithelium can't do sufficiently, as certain blood cells specialize to insert themselves between adjacent epithelial cells). The immunodepressive action was the initial normal behavior of the virus, similar to HIV. The fusion proteins were a way to spread the infection to other cells by simply merging them with the infected one (HIV does this too). It is believed that the ancestors of modern viviparous mammals evolved after an infection by this virus, enabling the fetus to survive the immune system of the mother.[24]
The human genome project found several thousand ERVs classified into 24 families.[25]
Immune network theory
A theoretical framework explaining the workings of the acquired immune system is provided by immune network theory, based on interactions between idiotypes (unique molecular features of one clonotype, i.e. the unique set of antigenic determinants of the variable portion of an antibody) and 'anti-idiotypes' (antigen receptors that react with the idiotype as if it were a foreign antigen). This theory, which builds on the existing clonal selection hypothesis and since 1974 has been developed mainly by Niels Jerne and Geoffrey W. Hoffmann, is seen as being relevant to the understanding of the HIV pathogenesis and the search for an HIV vaccine.
Stimulation of adaptive immunity
One of the most interesting developments in biomedical science during the past few decades has been elucidation of mechanisms mediating innate immunity. One set of innate immune mechanisms is humoral, such as complement activation. Another set comprises pattern recognition receptors such as toll-like receptors, which induce the production of interferons and other cytokines increasing resistance of cells such as monocytes to infections.[26] Cytokines produced during innate immune responses are among the activators of adaptive immune responses.[26] Antibodies exert additive or synergistic effects with mechanisms of innate immunity. Unstable HbS clusters Band-3, a major integral red cell protein;[27] antibodies recognize these clusters and accelerate their removal by phagocytic cells. Clustered Band 3 proteins with attached antibodies activate complement, and complement C3 fragments are opsonins recognized by the CR1 complement receptor on phagocytic cells.[28]
A population study has shown that the protective effect of the sickle-cell trait against falciparum malaria involves the augmentation of acquired as well as innate immune responses to the malaria parasite, illustrating the expected transition from innate to acquired immunity.[29]
Repeated malaria infections strengthen acquired immunity and broaden its effects against parasites expressing different surface antigens. By school age most children have developed efficacious adaptive immunity against malaria. These observations raise questions about mechanisms that favor the survival of most children in Africa while allowing some to develop potentially lethal infections.
In malaria, as in other infections,[26] innate immune responses lead into, and stimulate, adaptive immune responses. The genetic control of innate and acquired immunity is now a large and flourishing discipline.
Humoral and cell-mediated immune responses limit malaria parasite multiplication, and many cytokines contribute to the pathogenesis of malaria as well as to the resolution of infections.[30]
Evolution
The acquired immune system, which has been best-studied in mammals, originated in jawed fish approximately 500 million years ago. Most of the molecules, cells, tissues, and associated mechanisms of this system of defense are found in cartilaginous fishes.[31] Lymphocyte receptors, Ig and TCR, are found in all jawed vertebrates. The most ancient Ig class, IgM, is membrane-bound and then secreted upon stimulation of cartilaginous fish B cells. Another isotype, shark IgW, is related to mammalian IgD. TCRs, both α/β and γ/δ, are found in all animals from gnathostomes to mammals. The organization of gene segments that undergo gene rearrangement differs in cartilaginous fishes, which have a cluster form as compared to the translocon form in bony fish to mammals. Like TCR and Ig, the MHC is found only in jawed vertebrates. Genes involved in antigen processing and presentation, as well as the class I and class II genes, are closely linked within the MHC of almost all studied species.
Lymphoid cells can be identified in some pre-vertebrate deuterostomes (i.e., sea urchins).[32] These bind antigen with pattern recognition receptors (PRRs) of the innate immune system. In jawless fishes, two subsets of lymphocytes use variable lymphocyte receptors (VLRs) for antigen binding.[33] Diversity is generated by a cytosine deaminase-mediated rearrangement of LRR-based DNA segments.[34] There is no evidence for the recombination-activating genes (RAGs) that rearrange Ig and TCR gene segments in jawed vertebrates.
The evolution of the AIS, based on Ig, TCR, and MHC molecules, is thought to have arisen from two major evolutionary events: the transfer of the RAG transposon (possibly of viral origin) and two whole genome duplications.[31] Though the molecules of the AIS are well-conserved, they are also rapidly evolving. Yet, a comparative approach finds that many features are quite uniform across taxa. All the major features of the AIS arose early and quickly. Jawless fishes have a different AIS that relies on gene rearrangement to generate diverse immune receptors with a functional dichotomy that parallels Ig and TCR molecules.[35] The innate immune system, which has an important role in AIS activation, is the most important defense system of invertebrates and plants.
Types of acquired immunity
Immunity can be acquired either actively or passively. Immunity is acquired actively when a person is exposed to foreign substances and the immune system responds. Passive immunity is when antibodies are transferred from one host to another. Both actively acquired and passively acquired immunity can be obtained by natural or artificial means.
- Naturally Acquired Active Immunity – when a person is naturally exposed to antigens, becomes ill, then recovers.
- Naturally Acquired Passive Immunity – involves a natural transfer of antibodies from a mother to her infant. The antibodies cross the woman's placenta to the fetus. Antibodies can also be transferred through breast milk with the secretions of colostrum.
- Artificially Acquired Active Immunity – is done by vaccination (introducing dead or weakened antigen to the host's cell).
- Artificially Acquired Passive Immunity – This involves the introduction of antibodies rather than antigens to the human body. These antibodies are from an animal or person who is already immune to the disease.
| Naturally acquired | Artificially acquired |
|---|---|
| Active – Antigen enters the body naturally | Active – Antigens are introduced in vaccines. |
| Passive – Antibodies pass from mother to fetus via placenta or infant via the mother's milk. | Passive – Preformed antibodies in immune serum are introduced by injection. |
See also
| Wikimedia Commons has media related to Adaptive immunity. |
- Affinity maturation
- Allelic exclusion
- Anergy
- Immune response
- Immune tolerance
- Immunosuppression
- Original antigenic sin
- Somatic hypermutation
- Polyclonal response
Notes and references
- Notes
- References
- 1 2 3 4 5 6 Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walters P (2002). Molecular Biology of the Cell (4th ed.). New York and London: Garland Science. ISBN 0-8153-3218-1.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 Janeway CA, Travers P, Walport M, Shlomchik MJ (2001). Immunobiology (5th ed.). New York and London: Garland Science. ISBN 0-8153-4101-6.
- ↑ The innate and adaptive immune systems. Institute for Quality and Efficiency in Health Care (IQWiG). 4 August 2016.
- ↑ "Microbiology and Immunology On-Line Textbook". University of South Carolina School of Medicine. Archived from the original on 2 September 2008.
- 1 2 3 4 5 6 7 8 9 10 11 Janeway CA, Travers P, Walport M, Shlomchik MJ (2005). Immunobiology (6th ed.). Garland Science. ISBN 0-443-07310-4.
- 1 2 3 "Understanding the Immune System How It Works" (PDF). NIH Publication No. 03-5423. National Institute of Allergy and Infectious Diseases National Cancer Institute, U.S. Department of Health and Human Services National Institutes of Health. September 2003. Archived from the original (PDF) on 2007-01-03.
- ↑ Spencer LA, Weller PF (2010). "Eosinophils and Th2 immunity: contemporary insights". Immunology and Cell Biology. 88 (3): 250–56. doi:10.1038/icb.2009.115. PMC 3589820. PMID 20065995.
- ↑ Szabo PA, Levitin HM, Miron M, Snyder ME, Senda T, Yuan J, et al. (October 2019). "Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease". Nature Communications. 10 (1): 4706. Bibcode:2019NatCo..10.4706S. doi:10.1038/s41467-019-12464-3. PMC 6797728. PMID 31624246.
- ↑ Magen A, Nie J, Ciucci T, Tamoutounour S, Zhao Y, Mehta M, et al. (December 2019). "+ T Cells". Cell Reports. 29 (10): 3019–3032.e6. doi:10.1016/j.celrep.2019.10.131. PMC 6934378. PMID 31801070.
- ↑ Weinstein JS, Hernandez SG, Craft J (May 2012). "T cells that promote B-Cell maturation in systemic autoimmunity". Immunological Reviews. 247 (1): 160–71. doi:10.1111/j.1600-065x.2012.01122.x. PMC 3334351. PMID 22500839.
- ↑ Flajnik, Martin F. (19 March 2018). "A cold-blooded view of adaptive immunity". Nature Reviews Immunology. 18 (7): 438–53. doi:10.1038/s41577-018-0003-9. PMC 6084782. PMID 29556016.
- ↑ Kasamatsu, Jun (January 2013). "Evolution of innate and adaptive immune systems in jawless vertebrates". Microbiology and Immunology. 57 (1): 1–12. doi:10.1111/j.1348-0421.2012.00500.x. ISSN 0385-5600. PMID 22924515.
- ↑ Alder MN, Rogozin IB, Iyer LM, Glazko GV, Cooper MD, Pancer Z (December 2005). "Diversity and function of adaptive immune receptors in a jawless vertebrate". Science. 310 (5756): 1970–73. Bibcode:2005Sci...310.1970A. doi:10.1126/science.1119420. PMID 16373579.
- ↑ Mikonranta L, Mappes J, Kaukoniitty M, Freitak D (March 2014). "Insect immunity: oral exposure to a bacterial pathogen elicits free radical response and protects from a recurring infection". Frontiers in Zoology. 11 (1): 23. doi:10.1186/1742-9994-11-23. PMC 3975449. PMID 24602309.
- ↑ Sadd BM, Schmid-Hempel P (June 2006). "Insect immunity shows specificity in protection upon secondary pathogen exposure". Current Biology. 16 (12): 1206–10. doi:10.1016/j.cub.2006.04.047. PMID 16782011. S2CID 14436004.
- ↑ Pham LN, Dionne MS, Shirasu-Hiza M, Schneider DS (March 2007). "A specific primed immune response in Drosophila is dependent on phagocytes". PLOS Pathogens. 3 (3): e26. doi:10.1371/journal.ppat.0030026. PMC 1817657. PMID 17352533.
- ↑ Hernández López J, Schuehly W, Crailsheim K, Riessberger-Gallé U (June 2014). "Trans-generational immune priming in honeybees". Proceedings. Biological Sciences. 281 (1785): 20140454. doi:10.1098/rspb.2014.0454. PMC 4024302. PMID 24789904.
- ↑ Roth O, Joop G, Eggert H, Hilbert J, Daniel J, Schmid-Hempel P, Kurtz J (March 2010). "Paternally derived immune priming for offspring in the red flour beetle, Tribolium castaneum". The Journal of Animal Ecology. 79 (2): 403–13. doi:10.1111/j.1365-2656.2009.01617.x. PMID 19840170.
- 1 2 Dong Y, Taylor HE, Dimopoulos G (July 2006). "AgDscam, a hypervariable immunoglobulin domain-containing receptor of the Anopheles gambiae innate immune system". PLOS Biology. 4 (7): e229. doi:10.1371/journal.pbio.0040229. PMC 1479700. PMID 16774454.
- ↑ Meki IK, Kariithi HM, Parker AG, Vreysen MJ, Ros VI, Vlak JM, van Oers MM, Abd-Alla AM (November 2018). "RNA interference-based antiviral immune response against the salivary gland hypertrophy virus in Glossina pallidipes". BMC Microbiology. 18 (Suppl 1): 170. doi:10.1186/s12866-018-1298-1. PMC 6251114. PMID 30470195.
- ↑ Rubio M, Maestro JL, Piulachs MD, Belles X (June 2018). "Conserved association of Argonaute 1 and 2 proteins with miRNA and siRNA pathways throughout insect evolution, from cockroaches to flies". Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 1861 (6): 554–60. doi:10.1016/j.bbagrm.2018.04.001. hdl:10261/168051. PMID 29656113.
- ↑ International Human Genome Sequencing Consortium (October 2004). "Finishing the euchromatic sequence of the human genome". Nature. 431 (7011): 931–45. Bibcode:2004Natur.431..931H. doi:10.1038/nature03001. PMID 15496913.
- ↑ Mi S, Lee X, Li X, Veldman GM, Finnerty H, Racie L, LaVallie E, Tang XY, Edouard P, Howes S, Keith JC, McCoy JM (February 2000). "Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis". Nature. 403 (6771): 785–89. Bibcode:2000Natur.403..785M. doi:10.1038/35001608. PMID 10693809. S2CID 4367889.
- ↑ Villarreal LP. "The Viruses That Make Us: A Role For Endogenous Retrovirus In The Evolution Of Placental Species". University of California, Irvine (lecture notes). Archived from the original on 2007-07-15. Retrieved 2008-02-03.
- ↑ Villarreal LP (Oct 2001). "Persisting Viruses Could Play Role in Driving Host Evolution". ASM News. Archived from the original on 2009-05-08.
- 1 2 3 Uematsu S, Akira S (May 2007). "Toll-like receptors and Type I interferons". The Journal of Biological Chemistry. 282 (21): 15319–23. doi:10.1074/jbc.R700009200. PMID 17395581.
- ↑ Kuross SA, Rank BH, Hebbel RP (April 1988). "Excess heme in sickle erythrocyte inside-out membranes: possible role in thiol oxidation" (PDF). Blood. 71 (4): 876–82. doi:10.1182/blood.V71.4.876.876. PMID 3355895.
- ↑ Arese P, Turrini F, Schwarzer E (2005). "Band 3/complement-mediated recognition and removal of normally senescent and pathological human erythrocytes". Cellular Physiology and Biochemistry. 16 (4–6): 133–46. doi:10.1159/000089839. PMID 16301814.
- ↑ Williams TN, Mwangi TW, Roberts DJ, Alexander ND, Weatherall DJ, Wambua S, Kortok M, Snow RW, Marsh K (May 2005). "An immune basis for malaria protection by the sickle cell trait". PLOS Medicine. 2 (5): e128. doi:10.1371/journal.pmed.0020128. PMC 1140945. PMID 15916466.
- ↑ Schofield L, Grau GE (September 2005). "Immunological processes in malaria pathogenesis". Nature Reviews. Immunology. 5 (9): 722–35. doi:10.1038/nri1686. PMID 16138104. S2CID 19594405.
- 1 2 Flajnik MF, Kasahara M (January 2010). "Origin and evolution of the adaptive immune system: genetic events and selective pressures". Nature Reviews. Genetics. 11 (1): 47–59. doi:10.1038/nrg2703. PMC 3805090. PMID 19997068.
- ↑ Hibino T, Loza-Coll M, Messier C, Majeske AJ, Cohen AH, Terwilliger DP, Buckley KM, Brockton V, Nair SV, Berney K, Fugmann SD, Anderson MK, Pancer Z, Cameron RA, Smith LC, Rast JP (December 2006). "The immune gene repertoire encoded in the purple sea urchin genome" (PDF). Developmental Biology. 300 (1): 349–65. doi:10.1016/j.ydbio.2006.08.065. PMID 17027739.
- ↑ Pancer Z, Amemiya CT, Ehrhardt GR, Ceitlin J, Gartland GL, Cooper MD (July 2004). "Somatic diversification of variable lymphocyte receptors in the agnathan sea lamprey" (PDF). Nature. 430 (6996): 174–80. Bibcode:2004Natur.430..174P. doi:10.1038/nature02740. hdl:2027.42/62870. PMID 15241406. S2CID 876413.
- ↑ Rogozin IB, Iyer LM, Liang L, Glazko GV, Liston VG, Pavlov YI, Aravind L, Pancer Z (June 2007). "Evolution and diversification of lamprey antigen receptors: evidence for involvement of an AID-APOBEC family cytosine deaminase". Nature Immunology. 8 (6): 647–56. doi:10.1038/ni1463. PMID 17468760. S2CID 3658963.
- ↑ Boehm T (May 2011). "Design principles of adaptive immune systems". Nature Reviews. Immunology. 11 (5): 307–17. doi:10.1038/nri2944. PMID 21475308. S2CID 25989912.