Mitochondrial DNA depletion syndrome
| Mitochondrial DNA depletion syndrome | |
|---|---|
| Other names: mtDNA depletion syndrome | |
 | |
| Mitochondrial DNA depletion syndrome is inherited in an autosomal recessive manner | |
Mitochondrial DNA depletion syndrome (MDS or MDDS), or Alper's disease, is any of a group of autosomal recessive disorders that cause a significant drop in mitochondrial DNA in affected tissues. Symptoms can be any combination of myopathic, hepatopathic, or encephalomyopathic.[1] These syndromes affect tissue in the muscle, liver, or both the muscle and brain, respectively. The condition is typically fatal in infancy and early childhood, though some have survived to their teenage years with the myopathic variant and some have survived into adulthood with the SUCLA2 encephalomyopathic variant.[2][3] There is currently no curative treatment for any form of MDDS, though some preliminary treatments have shown a reduction in symptoms.[4]
Signs and symptoms
All forms of MDDS are very rare. MDDS causes a wide range of symptoms, which can appear in newborns, infants, children, or adults, depending on the class of MDDS; within each class symptoms are also diverse.[5]
In MDDS associated with mutations in TK2, infants generally develop normally, but by around two years of age, symptoms of general muscle weakness (called "hypotonia"), tiredness, lack of stamina, and difficulty feeding begin to appear. Some toddlers start to lose control of the muscles in their face, mouth, and throat, and may have difficulty swallowing. Motor skills that had been learned may be lost, but generally the functioning of the brain and ability to think are not affected.[5]
In MDDS associated with mutations in SUCLA2 or SUCLG1 that primarily affect the brain and muscle, hypotonia generally arises in infants before they are 6 months old, their muscles begin wasting away, and there is delay in psychomotor learning (learning basic skills like walking, talking, and intentional, coordinated movement). The spine often begins to curve (scoliosis or kyphosis), and the child often has abnormal movements (dystonia, athetosis or chorea), difficulty feeding, acid reflux, hearing loss, stunted growth, and difficulty breathing that can lead to frequent lung infections. Sometime epilepsy develops.[5]
In MDDS associated with mutations in RRM2B that primarily affect the brain and muscle, there is again hypotonia in the first months, symptoms of lactic acidosis like nausea, vomiting, and rapid deep breathing, failure to thrive including the head remaining small, delay or regression in moving, and hearing loss. Many body systems are affected.[5][6] The Charlie Gard case was associated with this sub form of the disease.[7]
In MDDS associated with mutations in DGUOK that primarily affect the brain and the liver, there are two forms. There is an early-onset form in which symptoms arise from problems in many organs in the first week of life, especially symptoms of lactic acidosis as well as low blood sugar. Within weeks of birth they can develop liver failure and the associated jaundice and abdominal swelling, and many neurological problems including developmental delays and regression, and uncontrolled eye movement. Rarely within this class of already rare diseases, symptoms only relating to liver disease emerge later in infancy or in childhood.[5]
In MDDS associated with mutations in MPV17 that primarily affect the brain and the liver, the symptoms are similar to those caused by DGUOK and also emerge shortly after birth, generally with fewer and less severe neurological problems. There is a subset of people of Navajo descent who develop Navajo neurohepatopathy, who in addition to these symptoms also have easily broken bones that do not cause pain, deformed hands or feet, and problems with their corneas.[5]
In MDDS associated with mutations in POLG that primarily affect the brain and the liver,[8] the symptoms are very diverse and can emerge anytime from shortly after birth to old age. The first signs of the disease, which include intractable seizures and failure to meet meaningful developmental milestones, usually occur in infancy, after the first year of life, but sometimes as late as the fifth year. Primary symptoms of the disease are developmental delay, progressive intellectual disability, hypotonia (low muscle tone), spasticity (stiffness of the limbs) possibly leading to quadriplegia, and progressive dementia. Seizures may include epilepsia partialis continua, a type of seizure that consists of repeated myoclonic (muscle) jerks. Optic atrophy may also occur, often leading to blindness. Hearing loss may also occur. Additionally, although physical signs of chronic liver dysfunction may not be present, many people experience liver impairment leading to liver failure.[9][10]
In MDDS associated with mutations in PEO1/C10orf2 that primarily affect the brain and the liver, symptoms emerge shortly after birth or in early infancy, with hypotonia, symptoms of lactic acidosis, enlarged liver, feeding problems, lack of growth, and delay of psychomotor skills. Neurologically, development is slowed or stopped, and epilepsy emerges, as do sensory problems like loss of eye control and deafness, and neuromuscular problems like a lack of reflexes, muscular atrophy, and twitching, and epilepsy.[5]
In MDDS associated with mutations in the genes associated with mutations in ECGF1/TYMP that primarily affects the brain and the gastrointestinal tract, symptoms can emerge any time in the first fifty years of life; most often they emerge before the person turns 20. Weight loss is common as is a lack of the ability of the stomach and intestines to automatically expand and contract and thus move through it (called gastrointestinal motility) – this leads to feeling full after eating only small amounts of food, nausea, acid reflux, All affected individuals develop weight loss and progressive gastrointestinal dysmotility manifesting as early satiety, nausea, diarrhea, vomiting, and stomach pain and swelling. People also develop neuropathy, with weakness and tingling. There are often eye problems, and intellectual disability.[5]
Causes
MDDS is caused by mutations that may be inherited from the parents or may form spontaneously during development of the fetus.[5] MDDS is associated with mutations in the genes TK2, SUCLA2, RRM2B, DGUOK, POLG, TYMP, SUCLG1, and TWNK.
Myopathic MDS is strongly correlated to a variety of mutations in the gene TK2, seeing a reduction of TK2 activity to less than 32% in people with MDS found with the mutation. Because TK2 plays a key role in the mitochondrial salvage pathways of several deoxyribonucleoside triphosphates (dNTPs), a lowered activity would lead to less cycling of nucleotides. This lack of nucleotide recycling is detrimental since the mitochondria cannot synthesize entirely new deoxynucleotides, and the inner membrane of the mitochondria prevents the negatively charged nucleotides of the cytosol from entering.[11]
The SUCLA2 gene codes for the beta-subunit of SCS-A. This enzyme catalyzes the synthesis of succinate and coenzyme A into succinyl-CoA, but is also associated with the complex formed by nucleoside diphosphate kinase (NDPK) in the last step of the dNTP salvage pathway.[12]
The RRM2B gene, which is expressed in the cell nucleus, codes for one of two versions of the R2 subunit of ribonucleotide reductase, which generates nucleotide precursors required for DNA replication by reducing ribonucleoside diphosphates to deoxyribonucleoside diphosphates. The version of R2 encoded by RRM2B is induced by TP53, and is required for normal DNA repair and mtDNA synthesis in non-proliferating cells. The other form of R2 is expressed only in dividing cells.[13]
The DGUOK gene encodes for mitochondrial deoxyguanosine kinase (dGK), which catalyzes the phosphorylation of deoxyribonucleosides into nucleotides.[14] POLG encodes for the catalytic subunit pol γA, which is part of mitochondrial DNA polymerase.[15]
Other causes are mutations of thymidine phosphorylase (TyMP), succinate-CoA ligase, alpha sub unit (SUCLG1) and TWNK (also known as PEO1 and C10orf2).[3][16]
Diagnosis
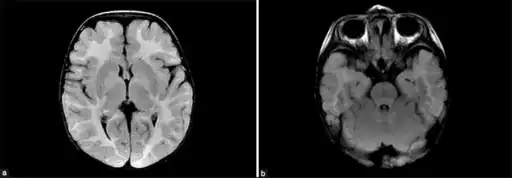
MDDS is diagnosed based on systemic symptoms presenting in infants, followed by a clinical examination and laboratory tests (for example, high lactate levels are common) medical imaging, and usually is finally confirmed and formally identified by genetic testing.[5]
Classification
MDDS are a group of genetic disorders that share a common pathology — a lack of functioning DNA in mitochondria.[5] There are generally four classes of MDDS:[5]
- a form that primarily affects muscle associated with mutations in the TK2 gene;
- a form that primarily affects the brain and muscle associated with mutations in the genes SUCLA2, SUCLG1, or RRM2B;
- a form that primarily affects the brain and the liver associated with mutations in DGUOK, MPV17, POLG, or TWNK (also called PEO1); and
- a form that primarily affects the brain and the gastrointestinal tract associated with mutations in ECGF1 (also called TYMP).
Treatment
There are no treatments for MDDS, but some of the symptoms can be managed. For survivors living with MDDS, there are drugs to control epilepsy, and physical therapy can help with muscle control. Liver transplants may benefit people with liver involvement.[5]
Prognosis
Myopathic form
The TK2 related myopathic form results in muscle weakness, rapidly progresses, leading to respiratory failure and death within a few years of onset. The most common cause of death is pulmonary infection. Only a few people have survived to late childhood and adolescence.[5]
Encephalomyopathic form
SUCLA2 and RRM2B related forms result in deformities to the brain.[5] A 2007 study based on 12 cases from the Faroe Islands (where there is a relatively high incidence due to a founder effect) suggested that the outcome is often poor with early lethality.[17] More recent studies (2015) with 50 people with SUCLA2 mutations, with range of 16 different mutations, show a high variability in outcomes with a number of people surviving into adulthood (median survival was 20 years). There is significant evidence (p = 0.020) that people with missense mutations have longer survival rates, which might mean that some of the resulting protein has some residual enzyme activity.[2]
RRM2B mutations have been reported in 16 infants with severe encephalomyopathic MDS that is associated with early-onset (neonatal or infantile), multi-organ presentation, and mortality during infancy.[5]
Hepatopathic form
DGUOK, POLG, and MPV17 related forms result in defects to the liver.[5] Liver dysfunction is progressive in the majority of individuals with both forms of DGUOK-related MDS and is the most common cause of death. For children with the multi-organ form, liver transplantation provides no survival benefit.[18]
Liver disease typically progresses to liver failure in affected children with MPV17-related MDS and liver transplantation remains the only treatment option for liver failure. Approximately half of affected children reported did not undergo liver transplantation and died because of progressive liver failure – the majority during infancy or early childhood. A few children were reported to survive without liver transplantation.[19]
Research
Nucleoside bypass therapy is an experimental treatment aimed to restore the normal levels of deoxyribonucleotides (dNTPs) in mitochondria.[5][20][21]
See also
- Charlie Gard case
References
- ↑ Elpeleg O (2003). "Inherited mitochondrial DNA depletion". Pediatr Res. 54 (2): 153–9. doi:10.1203/01.PDR.0000072796.25097.A5. PMID 12736387.
- 1 2 Carrozzo R, Verrigni D, Rasmussen M, de Coo R, Amartino H, Bianchi M, et al. (March 2016). "Succinate-CoA ligase deficiency due to mutations in SUCLA2 and SUCLG1: phenotype and genotype correlations in 71 patients". Journal of Inherited Metabolic Disease. 39 (2): 243–52. doi:10.1007/s10545-015-9894-9. PMID 26475597. S2CID 7881205.
- 1 2 Finsterer, J; Ahting, U (September 2013). "Mitochondrial depletion syndromes in children and adults". The Canadian Journal of Neurological Sciences. 40 (5): 635–44. doi:10.1017/S0317167100014852. PMID 23968935.
- ↑ Saito K, Kimura N, Oda N, Shimomura H, Kumada T, Miyajima T, Murayama K, Tanaka M, Fujii T (May 2012). "Pyruvate therapy for mitochondrial DNA depletion syndrome". Biochimica et Biophysica Acta (BBA) - General Subjects. 1820 (5): 632–6. doi:10.1016/j.bbagen.2011.08.006. PMID 21855607.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 El-Hattab AW, Scaglia F (April 2013). "Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options". Neurotherapeutics. 10 (2): 186–98. doi:10.1007/s13311-013-0177-6. PMC 3625391. PMID 23385875.
- ↑ Gorman, Gráinne S.; Taylor, Robert W. (April 17, 2014). "RRM2B-Related Mitochondrial Disease". GeneReviews. University of Washington, Seattle. PMID 24741716. Archived from the original on August 8, 2022. Retrieved October 21, 2022.
- ↑ Wheaton, Oliver (2017-07-25). "What is Charlie Gard's condition mitochondrial DNA depletion syndrome (or MDDS)?". Metro. Archived from the original on 2021-09-24. Retrieved 2021-09-24.
- ↑ This form of MDDS is also called "Alpers' disease", also called "Alpers' syndrome", "Alpers-Huttenlocher syndrome", "progressive sclerosing poliodystrophy", and "progressive infantile poliodystrophy". It is named after Bernard Jacob Alpers (Alpers' disease at Who Named It?) and Peter Huttenlocher (see Easton, John (19 August 2013). "Peter Huttenlocher, pediatric neurologist, 1931–2013". The University of Chicago. Archived from the original on 11 November 2013. Retrieved 1 November 2013.)
- ↑ "Alpers' Disease Information Page". National Institute of Neurological Disorders and Stroke. Archived from the original on 8 October 2019. Retrieved 24 July 2017.
- ↑ Cohen, BH; Chinnery, PF; Copeland, WC (December 18, 2014). Pagon, RA; et al. (eds.). "POLG-Related Disorders". GeneReviews. PMID 20301791.
- ↑ Saada A (December 2004). "Deoxyribonucleotides and disorders of mitochondrial DNA integrity". DNA and Cell Biology. 23 (12): 797–806. doi:10.1089/dna.2004.23.797. PMID 15684706. S2CID 20619194.
- ↑ Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, Bondi-Rubinstein G, Rahman S, Pagnamenta A, Eshhar S, Saada A (June 2005). "Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion". American Journal of Human Genetics. 76 (6): 1081–6. doi:10.1086/430843. PMC 1196446. PMID 15877282.
- ↑ Copeland, WC (2012). "Defects in mitochondrial DNA replication and human disease". Critical Reviews in Biochemistry and Molecular Biology. 47 (1): 64–74. doi:10.3109/10409238.2011.632763. PMC 3244805. PMID 22176657.
- ↑ Wang L, Limongelli A, Vila MR, Carrara F, Zeviani M, Eriksson S (January 2005). "Molecular insight into mitochondrial DNA depletion syndrome in two patients with novel mutations in the deoxyguanosine kinase and thymidine kinase 2 genes". Molecular Genetics and Metabolism. 84 (1): 75–82. doi:10.1016/j.ymgme.2004.09.005. PMID 15639197.
- ↑ Van Goethem G, Dermaut B, Löfgren A, Martin JJ, Van Broeckhoven C (July 2001). "Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions". Nature Genetics. 28 (3): 211–2. doi:10.1038/90034. PMID 11431686. S2CID 35417835.
- ↑ "Depletionssyndrom, mitochondriales (MDS)". Labor Lademannbogen (in Deutsch). Archived from the original on 2022-01-26. Retrieved 2022-10-21.
- ↑ Ostergaard E (18 May 2017). "SUCLA2-Related Mitochondrial DNA Depletion Syndrome, Encephalomyopathic Form with Methylmalonic Aciduria". In Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, Bird TD, Fong CT, Mefford HC, Smith RJ, Stephens K (eds.). SUCLA2-Related Mitochondrial DNA Depletion Syndrome, Encephalomyopathic Form, with Mild Methylmalonic Acuduria. GeneReviews [Internet]. University of Washington, Seattle. PMID 20301762. Archived from the original on 27 February 2021. Retrieved 8 July 2020 – via NCBI Bookshelf (National Library of Medicine).
- ↑ Dimmock DP, Dunn JK, Feigenbaum A, Rupar A, Horvath R, Freisinger P, Mousson de Camaret B, Wong LJ, Scaglia F (October 2008). "Abnormal neurological features predict poor survival and should preclude liver transplantation in patients with deoxyguanosine kinase deficiency". Liver Transplantation. 14 (10): 1480–5. doi:10.1002/lt.21556. PMID 18825706. S2CID 28819842.
- ↑ El-Hattab AW, Li FY, Schmitt E, Zhang S, Craigen WJ, Wong LJ (March 2010). "MPV17-associated hepatocerebral mitochondrial DNA depletion syndrome: new patients and novel mutations". Molecular Genetics and Metabolism. 99 (3): 300–8. doi:10.1016/j.ymgme.2009.10.003. PMID 20074988.
- ↑ Viscomi, Carlo; Bottani, Emanuela; Zeviani, Massimo (2015-06-01). "Emerging concepts in the therapy of mitochondrial disease". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1847 (6): 544–557. doi:10.1016/j.bbabio.2015.03.001. PMID 25766847.
- ↑ Cámara, Yolanda; González-Vioque, Emiliano; Scarpelli, Mauro; Torres-Torronteras, Javier; Martí, Ramon (2013-10-01). "Feeding the deoxyribonucleoside salvage pathway to rescue mitochondrial DNA". Drug Discovery Today. 18 (19): 950–957. doi:10.1016/j.drudis.2013.06.009. PMID 23817075.
External links
| Classification | |
|---|---|
| External resources |
|