Krabbe disease
| Krabbe disease | |
|---|---|
Other names:
| |
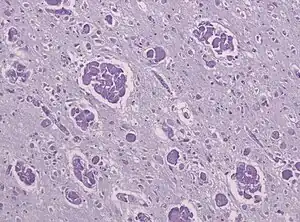 | |
| A histopathology slide of a brain with Krabbe disease showing giant cells with PAS stain inclusions ("globoid cells") within astrocytic gliosis and loss of myelinated fibers. | |
| Symptoms |
|
| Usual onset | Within 3 to 6 months of birth, but can present in childhood or even adulthood |
| Types | Infantile, juvenile and adult |
| Causes | Mutation of GALC gene |
| Risk factors | Parents who are heterozygous (only one copy) for the mutation to the GALC gene |
| Diagnostic method | Histopathology, genetic testing[1] |
| Prevention | Prenatal diagnosis and screening of at-risk couples[1] |
| Treatment | Symptomatic and supportive treatment only, but stem cell transplantation may be beneficial[1] |
| Prognosis | One-, two-, and three-year survival rates of 60%, 26%, and 14%, respectively[2] |
Krabbe disease (KD), also known as globoid cell leukodystrophy[3] and galactosylceramide lipidosis, is a rare and often fatal lysosomal storage disease that results in progressive damage to the nervous system. KD involves dysfunctional metabolism of sphingolipids and is inherited in an autosomal recessive pattern. The disease is named after the Danish neurologist Knud Krabbe (1885–1961).[4]
Signs and symptoms
Symptoms in asymptomatic infantile-onset (<12 months after birth) and later-onset Krabbe disease present themselves differently. Of individuals with infantile-onset Krabbe disease, 85–90% display progressive neurologic deterioration in infancy and death before the age of two.[5] Symptoms include irritability, fevers, limb stiffness, seizures, feeding difficulties (like GERD), vomiting, staring episodes, and slowing of mental and motor development. In the first stages of the disease, doctors often mistake the symptoms for those of cerebral palsy. Other symptoms include muscle weakness, spasticity, deafness, optic atrophy, optic nerve enlargement,[6] blindness, paralysis, and difficulty when swallowing. Prolonged weight loss may also occur.
10–15% of individuals with later-onset Krabbe disease have a much slower disease progression. These individuals may also display symptoms such as esotropia, slurred speech, and slow development or loss of motor milestones.[5]
Causes

Krabbe disease is caused by mutations in the GALC gene located on chromosome 14 (14q31),[7] which is inherited in an autosomal recessive manner. Mutations in the GALC gene cause a deficiency of an enzyme called galactosylceramidase.[8] In rare cases, it may be caused by a lack of active saposin A (a derivative of prosaposin).[1]
The buildup of unmetabolized lipids adversely affects the growth of the nerve's protective myelin sheath (the covering that insulates many nerves) resulting in demyelination and severe progressive degeneration of motor skills. As part of a group of disorders known as leukodystrophies, Krabbe disease results from the imperfect growth and development of myelin.
Galactosylceramidase deficiency also results in a buildup of a glycosphingolipid called psychosine, which is toxic to oligodendrocytes, a type of non-neuronal cell found in the nervous system, collectively termed neuroglia.[9]
Diagnosis
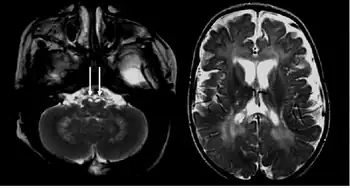
There are a few ways to help pinpoint the presence of Krabbe disease. Newborn screening for Krabbe disease includes assaying dried blood cells for GALC enzyme activity and molecular analysis for evidence of GALC enzyme mutations. Infants displaying low enzyme activity and/or enzyme mutations should be referred for additional diagnostic testing and neurological examination.[10] 0-5% GALC enzyme activity is observed in all symptomatic individuals with Krabbe disease.[5] High concentration of psychosine in dried blood spots may also be identified as a marker for Krabbe disease.[11] A 2011 study discovered that individuals with Krabbe disease, more so in later-onset individuals, tend to have an abnormal increase in CSF protein concentration.[12]
The disease may be diagnosed by its characteristic grouping of certain cells (multinucleated globoid cells), nerve demyelination and degeneration, and destruction of brain cells. Special stains for myelin (e.g., luxol fast blue) may be used to aid diagnosis.
New York,[13] Missouri and Kentucky[14] include Krabbe in the newborn screening panel.[15] Indiana will start screening in 2020.[16] Parents of newborns with a diagnosis of Krabbe disease can access online community support and resources below.
Treatment
Although there is no known cure for Krabbe disease, bone marrow transplantation or hematopoietic stem cell transplantation (HSCT) has been shown to benefit cases early in the course of the disease. Generally, treatment for the disorder is symptomatic and supportive. Physical therapy may help maintain or increase muscle tone and circulation.
A 15-year study on the developmental outcomes of children with Krabbe disease who underwent HSCT in the first seven weeks after birth found that patients have a better prognosis for both lifespan and functionality, with a slower progression of the disease.[17] Even symptomatic individuals with later-onset Krabbe disease may benefit from HSCT if diagnosed early enough.[18] Umbilical-cord blood is typically used as the source for the transplant stem cells.[19] Clinical trials for gene therapy are currently enrolling patients.[20]
Management
Symptom management can be particularly difficult for individuals with infantile onset, as symptoms tend to progress rapidly.[19] Because there is no treatment for Krabbe disease, management of the condition is typically supportive and aimed at alleviating symptoms. Frequent evaluation is encouraged in order to anticipate the onset of, and preparation for, certain symptoms.[5] Physical therapy can help to alleviate motor difficulties and increase strength, mobility, and flexibility.[5]
Gastrostomy tubes are used to circumvent feeding difficulties and prevent aspiration. A simultaneous gastrostomy tube insertion and Nissen fundoplication procedure is commonly performed to prevent the need for a secondary surgical procedure.[19] Individuals with Krabbe disease with severe motor deficits tend to be more susceptible to overfeeding, as they require less calorie consumption and thus consume fewer calories than what caretakers may expect.[19] There is also evidence that routine vaccines may accelerate disease progression; many individuals with Krabbe disease tend to not follow traditional vaccination procedures.[19]
Prognosis
In infantile Krabbe disease, death usually occurs in early childhood. A 2011 study found one-, two-, and three-year survival rates of 60%, 26%, and 14%, respectively, with a few surviving longer. Patients with late-onset Krabbe disease tend to have a slower progression of the disease and live significantly longer.[2]
Epidemiology
This disease does not only impact humans, but other animals such as monkeys, mice, and dogs have been observed to develop Krabbe disease as well. While certain gene deletions are more frequent than others, novel mutations resulting in Krabbe disease have been discovered worldwide. Most commonly, the underlying cause of the disease is a deletion of a GALC gene, which causes a deficiency in the GALC enzyme. This is the circumstance in 80% of patients who have European and Mexican origins.[21] The mortality rate of early infantile Krabbe disease is 90% before the age of two. Later onset of symptoms is associated with longer life expectancy, with older children generally surviving two to seven years after the initial diagnosis.[22]
Krabbe disease occurs in about one in 100,000 births.[23] Because the disease is genetic, incidence rates vary widely from population to population.[21] The incidence rate is extremely low in Japan, with between 5 and 10 cases per 1,000,000 live births. In the United States, Krabbe disease occurs in approximately 1 out of every 100,000 live births.[24] Scandinavian countries report incidence rates of one in 50,000 births.[25] In certain communities Krabbe disease is much more frequent, such as the Druze community in Israel, which has an incidence rate of 6 out of every 1,000 live births.[24] This higher rate is thought to be due in part to a high frequency of consanguineous marriages. Almost 35% of all Druze marriages were found to be between first-cousin familial relations.[26] There have been no reported cases of Krabbe disease among the Jewish community.[24]
Time of onset also varies in frequency by location. Early infantile Krabbe Disease is the most common form of the disease overall, but Nordic communities tend to have even higher rates of early infantile onset Krabbe disease, while Southern European countries have higher incidences of late-onset cases. It is difficult to estimate the incidence of adult-onset Krabbe disease, due to discrepancies in classifying cases late-onset versus adult-onset.[24]
Society and culture
Former Buffalo Bills quarterback Jim Kelly has been a leader in gaining recognition and research funding for Krabbe disease following the diagnosis of his son, Hunter, in 1997. Hunter Kelly died of the disease on August 5, 2005, at the age of eight. They created Hunter's Hope Archived 2020-12-08 at the Wayback Machine - a foundation that seeks to advance Newborn Screening, research and treatments, and provides support to families of leukodystrophy children.
Family advocacy is a critical part of advancing newborn screening, and many Krabbe families have made significant advocacy progress in their states.
As an example, Cove Ellis is a child from Georgia, United States who was diagnosed with the disease in early 2016. Ellis' family, along with her community, has worked to raise awareness of the disease and helped pass "Cove's Law", which provides parents the option to have prenatal screening for the disease, which can, potentially, save the child from the morbidity and mortality of Krabbe disease.[27]
Animals
Krabbe disease is found in mice[28] may also be found in cats[29] and in dogs, particularly the West Highland White Terriers and Cairn Terriers.[30][31]
See also
- Maria Luisa Escolar
- The Myelin Project
- The Stennis Foundation
- KrabbeConnect Archived 2022-06-15 at the Wayback Machine
- The Legacy of Angels Foundation Archived 2022-05-13 at the Wayback Machine
- Hunter's Hope Foundation Archived 2022-05-19 at the Wayback Machine
References
- 1 2 3 4 5 Langan, Thomas J (23 November 2016). "Krabbe disease". UpToDate. Archived from the original on 3 May 2017. Retrieved 18 October 2018.
- 1 2 Duffner, Patricia K.; Barczykowski, Amy; Jalal, Kabir; Yan, Li; Kay, Denise M.; Carter, Randy L. (September 2011). "Early Infantile Krabbe Disease: Results of the World-Wide Krabbe Registry". Pediatric Neurology. 45 (3): 141–148. doi:10.1016/j.pediatrneurol.2011.05.007. PMID 21824559.
- ↑ Li, Y; Sands, MS (November 2014). "Experimental therapies in the murine model of globoid cell leukodystrophy". Pediatric Neurology (Review). 51 (5): 600–6. doi:10.1016/j.pediatrneurol.2014.08.003. PMC 4252788. PMID 25240259.
- ↑ synd/1457 at Who Named It?
- 1 2 3 4 5 Orsini, Joseph J.; Escolar, Maria L.; Wasserstein, Melissa P.; Caggana, Michele (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Krabbe Disease", GeneReviews®, University of Washington, Seattle, PMID 20301416, archived from the original on 2017-01-18, retrieved 2019-11-25
- ↑ Hussain, S. A.; Zimmerman, H. H.; Abdul-Rahman, O. A.; Hussaini, S. M.; Parker, C. C.; Khan, M. (May 2011). "Optic Nerve Enlargement in Krabbe Disease: A Pathophysiologic and Clinical Perspective". Journal of Child Neurology. 26 (5): 642–644. doi:10.1177/0883073810387929. PMID 21285037. S2CID 22242663.
- ↑ Cannizzaro, L.A. (1994). "Regional mapping of the human galactocerebrosidase gene (GALC) to 14q31 by in situ hybridization". Cytogenetic and Genome Research. 66 (4): 244–245. doi:10.1159/000133703. PMID 8162701.
- ↑ "Krabbe disease". National Institutes of Health. Archived from the original on 2020-10-01. Retrieved 2022-05-21.
- ↑ Kohlschütter, A (2013). Lysosomal leukodystrophies: Krabbe disease and metachromatic leukodystrophy. Handbook of Clinical Neurology. Vol. 113. pp. 1611–18. doi:10.1016/B978-0-444-59565-2.00029-0. ISBN 9780444595652. PMID 23622382.
- ↑ Orsini, Joseph J.; Kay, Denise M.; Saavedra-Matiz, Carlos A.; Wenger, David A.; Duffner, Patricia K.; Erbe, Richard W.; Biski, Chad; Martin, Monica; Krein, Lea M.; Nichols, Matthew; Kurtzberg, Joanne (2016-03-01). "Newborn screening for Krabbe disease in New York State: the first eight years' experience". Genetics in Medicine. 18 (3): 239–248. doi:10.1038/gim.2015.211. ISSN 1530-0366. PMID 26795590.
- ↑ Escolar, ML; Kiely, BT; Shawgo, E; Hong, X; Gelb, MH; Orsini, JJ; Matern, D; Poe, MD (July 2017). "Psychosine, a marker of Krabbe phenotype and treatment effect". Molecular Genetics and Metabolism. 121 (3): 271–278. doi:10.1016/j.ymgme.2017.05.015. ISSN 1096-7192. PMC 5548593. PMID 28579020.
- ↑ Shah, Samir S.; Ebberson, Jessica; Kestenbaum, Lori A.; Hodinka, Richard L.; Zorc, Joseph J. (January 2011). "Age-Specific Reference Values for Cerebrospinal Fluid Protein Concentration in Neonates and Young Infants". Journal of Hospital Medicine. 6 (1): 22–27. doi:10.1002/jhm.711. ISSN 1553-5592. PMC 2978786. PMID 20629018.
- ↑ Duffner, Patricia K.; Caggana, Michele; Orsini, Joseph J.; Wenger, David A.; Patterson, Marc C.; Crosley, Carl J.; Kurtzberg, Joanne; Arnold, Georgianne L.; Escolar, Maria L. (2009-04-01). "Newborn Screening for Krabbe Disease: the New York State Model". Pediatric Neurology. 40 (4): 245–252. doi:10.1016/j.pediatrneurol.2008.11.010. PMID 19302934.
- ↑ (KRS 214.155)
- ↑ "unbs_state - Hunter's Hope Foundation". www.huntershope.org. Archived from the original on 2016-11-14. Retrieved 2016-11-14.
- ↑ Runevitch, Jennie. "Bryce's Battle: Family gets law change after deadly diagnosis". WTHR. Archived from the original on 2019-12-10. Retrieved 2022-05-21.
- ↑ Wright, Matthew D.; Poe, Michele D.; DeRenzo, Anthony; Haldal, Shilpa; Escolar, Maria L. (2017-09-26). "Developmental outcomes of cord blood transplantation for Krabbe disease: A 15-year study". Neurology. 89 (13): 1365–1372. doi:10.1212/WNL.0000000000004418. ISSN 1526-632X. PMC 5649761. PMID 28855403.
- ↑ Laule, Cornelia; Vavasour, Irene M.; Shahinfard, Elham; Mädler, Burkhard; Zhang, Jing; Li, David K. B.; MacKay, Alex L.; Sirrs, Sandra M. (May 2018). "Hematopoietic Stem Cell Transplantation in Late-Onset Krabbe Disease: No Evidence of Worsening Demyelination and Axonal Loss 4 Years Post-allograft". Journal of Neuroimaging. 28 (3): 252–255. doi:10.1111/jon.12502. ISSN 1552-6569. PMID 29479774. S2CID 3533589.
- 1 2 3 4 5 Escolar, Maria L.; West, Tara; Dallavecchia, Alessandra; Poe, Michele D.; LaPoint, Kathleen (November 2016). "Clinical management of Krabbe disease". Journal of Neuroscience Research. 94 (11): 1118–1125. doi:10.1002/jnr.23891. ISSN 1097-4547. PMID 27638597. S2CID 34083553.
- ↑ "A Phase 1/2 Clinical Study of Intravenous Gene Transfer With an AAVrh10 Vector Expressing GALC in Krabbe Subjects Receiving Hematopoietic Stem Cell Transplantation (RESKUE)". clinicaltrials.gov. 5 May 2021. Archived from the original on 10 March 2022. Retrieved 21 May 2022.
- 1 2 Amin, Mutaz; Elsayad, Liena; Ahmed, Ammar Eltahir (2017). "Clinical and Genetic Characteristics of Leukodystrophies in Africa". Journal of Neurosciences in Rural Practice. 8 (S 01): S089–S093. doi:10.4103/jnrp.jnrp_511_16. PMC 5602269. PMID 28936078.
- ↑ Mayo Clinic Staff (June 2018). "Krabbe Disease". Mayo Clinic. Archived from the original on 2019-07-27. Retrieved 2022-05-21.
- ↑ "Krabbe disease". Genetics Home Reference. United States National Library of Medicine. 2008-05-02. Archived from the original on 2012-08-16. Retrieved 2008-05-07.
- 1 2 3 4 Matsuda, Junko; Suzuki, Kunihiko (2007), Barranger, John A.; Cabrera-Salazar, Mario A. (eds.), "Krabbe Disease (Globoid Cell Leukodystrophy)", Lysosomal Storage Disorders, Springer US, pp. 269–283, doi:10.1007/978-0-387-70909-3_18, ISBN 9780387709093
- ↑ "Books.Google.com". Archived from the original on 2022-05-21. Retrieved 2022-05-21.
- ↑ Zayed, Hatem (February 2015). "Krabbe Disease in the Arab World" (PDF). Journal of Pediatric Genetics. 4 (2146–4596): 001–008. doi:10.1055/s-0035-1554981. PMC 4906415. PMID 27617109. Archived (PDF) from the original on 2019-12-06. Retrieved 2022-05-21.
- ↑ Miller, Andy (March 18, 2017). "Georgia lawmakers considering bill on testing newborns for rare genetic disorder". Athens Banner-Herald. Athens Banner-Herald. Archived from the original on 2019-04-05. Retrieved 25 March 2017.
- ↑ Suzuki, K. (July 1995). "The twitcher mouse: a model for Krabbe disease and for experimental therapies". Brain Pathology (Zurich, Switzerland). 5 (3): 249–258. doi:10.1111/j.1750-3639.1995.tb00601.x. ISSN 1015-6305. PMID 8520724. Archived from the original on 2022-03-24. Retrieved 2022-05-21.
- ↑ Salvadori C, Modenato M, Corlazzoli DS, Arispici M, Cantile C (May 2005). "Clinicopathological features of globoid cell leucodystrophy in cats". J. Comp. Pathol. 132 (4): 350–6. doi:10.1016/j.jcpa.2004.12.001. PMC 7172685. PMID 15893994.
- ↑ "NYtimes.com". Archived from the original on 2022-05-21. Retrieved 2022-05-21.
- ↑ Capucchio MT, Prunotto M, Lotti D, Valazza A, Galloni M, Dore B, Pregel P, Amedeo S, Catalano D, Cornaglia E, Schiffer D (2008). "Krabbe's disease in two West Highland White terriers". Clin. Neuropathol. 27 (5): 295–301. doi:10.5414/npp27295. PMID 18808060.
This article incorporates public domain text from the United States National Library of Medicine and the National Institute of Neurological Disorders and Stroke.
External links
- GeneReviews/NCBI/NIH/UW entry on Krabbe disease Archived 2017-01-18 at the Wayback Machine
- OMIM entries on Krabbe disease Archived 2010-06-04 at the Wayback Machine
| Classification | |
|---|---|
| External resources |
|