Niemann–Pick disease, SMPD1-associated
| Niemann–Pick disease, SMPD1-associated | |
|---|---|
| Other names: Niemann–Pick disease, SMPD1-associated | |
| Specialty | Endocrinology |
Acid sphingomyelinase deficiency (ASMD), or Niemann–Pick disease, SMPD1-associated, refers to two different types of Niemann–Pick disease which are associated with the SMPD1 gene.
There are approximately 1,200 cases of NPA and NPB worldwide with the majority of cases being Type B or an intermediate form.
Descriptions of type E[1] and type F[2] have been published, but they are not well characterized, and are currently classified under type B.[3]
Signs and symptoms
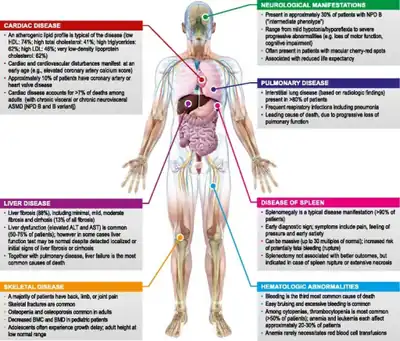
The clinical presentation of Acid sphingomyelinase deficiency is as follows:[4]
- Hepatosplenomegaly
- Neurologic deterioration
- Respiratory infection
Genetics
Mutations in the SMPD1 gene cause Niemann–Pick Types A and B. This gene carries instructions for cells to produce a lysosomal enzyme called acid sphingomyelinase. Insufficient activity of the enzyme acid sphingomyelinase causes the buildup of toxic amounts of sphingomyelin, a fatty substance present in every cell of the body. This enzyme is found in special compartments within cells called lysosomes (compartments that digest and recycle materials in the cell), and is required to metabolize the lipid sphingomyelin. If sphingomyelinase is absent or not functioning properly, sphingomyelin cannot be metabolized properly and is accumulated within the cell, eventually causing cell death and the malfunction of major organ systems.
Diagnosis
Type A
Niemann–Pick Type A, the most common type, occurs in infants and is characterized by jaundice, an enlarged liver, failure to thrive, progressive deterioration of the nervous system and profound brain damage. Children affected by Niemann Pick Type A rarely live beyond 18 months. Niemann–Pick Type A occurs more frequently among individuals of Ashkenazi (eastern and central European) Jewish descent than in other ethnicities. The incidence within the Ashkenazi population is approximately 1 in 40,000 people. The incidence for other populations is 1 in 250,000 people.
Type B
Niemann–Pick Type B involves an enlarged liver and spleen hepatosplenomegaly, growth retardation, and problems with lung function including frequent lung infections. Other signs include blood abnormalities such as abnormal cholesterol and lipid levels, and low numbers of blood cells involved in clotting (platelets). The brain is not affected in Type B and the disease often presents in the pre-teen years.
Treatment
In terms of the management of Acid sphingomyelinase deficiency the following is done:[4]
- Feeding therapy
- Blood transfusion
- Hepatic surgery (transplant)
- Physical therapy
- Occupational therapy
References
- ↑ Lynn R, Terry RD (December 1964). "Lipid histochemistry and electron microscopy in adult Niemann–Pick disease". The American Journal of Medicine. 37 (6): 987–94. doi:10.1016/0002-9343(64)90139-1. PMID 14246098.
- ↑ Schneider EL, Pentchev PG, Hibbert SR, Sawitsky A, Brady RO (October 1978). "A new form of Niemann–Pick disease characterised by temperature-labile sphingomyelinase". Journal of Medical Genetics. 15 (5): 370–4. doi:10.1136/jmg.15.5.370. PMC 1013734. PMID 216805.
- ↑ Online Mendelian Inheritance in Man (OMIM): Niemann–Pick Disease, Type B - 607616
- 1 2 Wasserstein, Melissa P.; Schuchman, Edward H. (1993). "Acid Sphingomyelinase Deficiency". GeneReviews®. University of Washington, Seattle. Archived from the original on 28 November 2022. Retrieved 2 January 2023.
External links
| Classification | |
|---|---|
| External resources |
|