Cori cycle
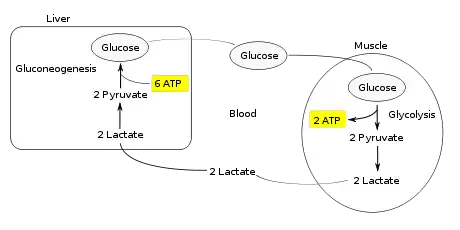
The Cori cycle (also known as the lactic acid cycle), named after its discoverers, Carl Ferdinand Cori and Gerty Cori,[1] is a metabolic pathway in which lactate produced by anaerobic glycolysis in muscles is transported to the liver and converted to glucose, which then returns to the muscles and is cyclically metabolized back to lactate.[2]
Process
_and_Carl_Ferdinand_Cori_-_restoration1.jpg.webp)
Muscular activity requires ATP, which is provided by the breakdown of glycogen in the skeletal muscles. The breakdown of glycogen, known as glycogenolysis, releases glucose in the form of glucose 1-phosphate (G1P). The G1P is converted to G6P by phosphoglucomutase. G6P is readily fed into glycolysis, (or can go into the pentose phosphate pathway if G6P concentration is high) a process that provides ATP to the muscle cells as an energy source. During muscular activity, the store of ATP needs to be constantly replenished. When the supply of oxygen is sufficient, this energy comes from feeding pyruvate, one product of glycolysis, into the citric acid cycle, which ultimately generates ATP through oxygen-dependent oxidative phosphorylation.
When oxygen supply is insufficient, typically during intense muscular activity, energy must be released through anaerobic metabolism. Lactic acid fermentation converts pyruvate to lactate by lactate dehydrogenase. Most importantly, fermentation regenerates NAD+, maintaining its concentration so additional glycolysis reactions can occur. The fermentation step oxidizes the NADH produced by glycolysis back to NAD+, transferring two electrons from NADH to reduce pyruvate into lactate. (Refer to the main articles on glycolysis and fermentation for the details.)
Instead of accumulating inside the muscle cells, lactate produced by anaerobic fermentation is taken up by the liver. This initiates the other half of the Cori cycle. In the liver, gluconeogenesis occurs. From an intuitive perspective, gluconeogenesis reverses both glycolysis and fermentation by converting lactate first into pyruvate, and finally back to glucose. The glucose is then supplied to the muscles through the bloodstream; it is ready to be fed into further glycolysis reactions. If muscle activity has stopped, the glucose is used to replenish the supplies of glycogen through glycogenesis.[3]
Overall, the glycolysis steps of the cycle produce 2 ATP molecules at a cost of 6 ATP molecules consumed in the gluconeogenesis steps. Each iteration of the cycle must be maintained by a net consumption of 4 ATP molecules. As a result, the cycle cannot be sustained indefinitely. The intensive consumption of ATP molecules in the Cori cycle shifts the metabolic burden from the muscles to the liver.
Significance
The cycle's importance is based on preventing lactic acidosis during anaerobic conditions in the muscle. However, normally, before this happens, the lactic acid is moved out of the muscles and into the liver.[3]
Additionally, this cycle is important in ATP production, an energy source, during muscle exertion. The end of muscle exertion allows the Cori cycle to function more effectively. This repays the oxygen debt so both the electron transport chain and citric acid cycle can produce energy at optimum effectiveness.[3] This acid attributes to the sore feeling in muscles after extensive exercising.
The Cori cycle is a much more important source of substrate for gluconeogenesis than food.[4][5] The contribution of Cori cycle lactate to overall glucose production increases with fasting duration before plateauing.[6] Specifically, after 12, 20, and 40 hours of fasting by human volunteers, gluconeogenesis accounts for 41%, 71%, and 92% of glucose production, but the contribution of Cori cycle lactate to gluconeogenesis is 18%, 35%, and 36%, respectively.[6] The remaining glucose production comes from protein breakdown,[6] muscle glycogen,[6] and glycerol from lipolysis.[7]
The drug metformin can cause lactic acidosis in patients with kidney failure because metformin inhibits the hepatic gluconeogenesis of the Cori cycle, particularly the mitochondrial respiratory chain complex 1.[8] The buildup of lactate and its substrates for lactate production, pyruvate and alanine, lead to excess lactate.[9] Normally, the excess acid that is the result of the inhibition of the mitochondrial chain complex would be cleared by the kidneys, but in patients with kidney failure, the kidneys cannot handle the excess acid. A common misconception posits that lactate is the agent responsible for the acidosis, but lactate is a conjugate base, being mostly ionised at physiologic pH, and serves as a marker of associated acid production rather than being its cause. [10] [11]
See also
- Alanine cycle
- Citric acid cycle
References
- ↑ "Carl and Gerty Cori and Carbohydrate Metabolism". National Historic Chemical Landmark. American Chemical Society. 2004. Retrieved 12 May 2020.
- ↑ Nelson DL, Cox MM (2005). Lehninger Principles of Biochemistry (Fourth ed.). New York: W.H. Freeman and Company. p. 543. ISBN 978-0-7167-4339-2.
- 1 2 3 "Ophardt CE (2003). "Cori Cycle". Virtual Chem Book. Elmhurst College. pp. 1–3. Archived from the original on 23 April 2008. Retrieved 3 May 2008.
- ↑ Gerich JE, Meyer C, Woerle HJ, Stumvoll M (February 2001). "Renal gluconeogenesis: its importance in human glucose homeostasis". Diabetes Care. 24 (2): 382–91. doi:10.2337/diacare.24.2.382. PMID 11213896.
- ↑ Nuttall FQ, Ngo A, Gannon MC (September 2008). "Regulation of hepatic glucose production and the role of gluconeogenesis in humans: is the rate of gluconeogenesis constant?". Diabetes/Metabolism Research and Reviews. 24 (6): 438–58. doi:10.1002/dmrr.863. PMID 18561209. S2CID 24330397.
- 1 2 3 4 Katz J, Tayek JA (September 1998). "Gluconeogenesis and the Cori cycle in 12-, 20-, and 40-h-fasted humans". The American Journal of Physiology. 275 (3): E537-42. doi:10.1152/ajpendo.1998.275.3.E537. PMID 9725823.
- ↑ Cahill GF (2006). "Fuel metabolism in starvation". Annual Review of Nutrition. 26: 1–22. doi:10.1146/annurev.nutr.26.061505.111258. PMID 16848698.
- ↑ Vecchio S, Giampreti A, Petrolini VM, Lonati D, Protti A, Papa P, et al. (February 2014). "Metformin accumulation: lactic acidosis and high plasmatic metformin levels in a retrospective case series of 66 patients on chronic therapy". Clinical Toxicology. 52 (2): 129–35. doi:10.3109/15563650.2013.860985. PMID 24283301. S2CID 23259898.
- ↑ Sirtori CR, Pasik C (1994). "Re-evaluation of a biguanide, metformin: mechanism of action and tolerability". Pharmacological Research. 30 (3): 187–228. doi:10.1016/1043-6618(94)80104-5. PMID 7862618.
- ↑ "The myth of lactic acidosis".
- ↑ "Metformin toxicity".
Further reading
- Smith AD, Datta SP, Smith GH, Campbell PN, Bentley R, eds. (1997). Oxford Dictionary of Biochemistry and Molecular Biology. New York: Oxford University Press. ISBN 978-0-19-854768-6.