Cri du chat syndrome
| Cri du chat syndrome | |
|---|---|
| Other names: Chromosome 5p deletion syndrome, 5p− syndrome, cat cry syndrome, Lejeune syndrome[1][2] | |
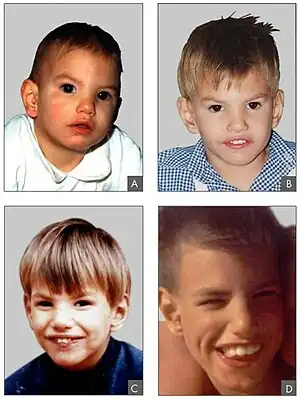 | |
| Facial features in Cri du chat syndrome at the age of 8 months (A), 2 years (B), 4 years (C) and 9 years (D)[3] | |
| Specialty | Medical genetics |
| Symptoms | High pitched cry, small head, specific facial features[1] |
| Complications | Trouble breathing, trouble feeding, behavioral problems, intellectual disability[1] |
| Usual onset | Present at birth[1] |
| Causes | Unknown[4] |
| Diagnostic method | Suspected based on symptoms, confirmed by genetic testing[1] |
| Differential diagnosis | Wolf-Hirschhorn syndrome[2] |
| Treatment | Based on symptoms[1] |
| Prognosis | Typically normal life expectancy[4] |
| Frequency | ~ 1 in 30,000 newborns[1] |
Cri du chat syndrome is a genetic disorder due to a partial deletion of genetic material on chromosome 5.[4] Symptoms often include a high pitched cry, small head, and specific facial features.[1] Complications may include trouble breathing, trouble feeding, behavioral problems, and intellectual disability.[1]
The cause is unknown.[4] The loss of genetic material that results in this syndrome generally occurs during early development.[1] In about a 10th of cases it is inherited from an unaffected parent.[1] Diagnosis is suspected based on symptoms and confirmed by genetic testing.[1]
Treatment is directed at the symptoms.[1] This may include special education, physical therapy, and speech therapy.[2] Some individuals learn short sentence while others express themselves with gestures.[4] Generally they are happy and friendly.[4] Most of those affected have a normal life expectancy.[4]
The condition affects an estimated 1 in 15,000 to 1 in 50,000 babies.[1] It affects females more often than males.[2] It was first described by Jérôme Lejeune in 1963.[2][5] Its name is the French term for "cat-cry".[2]
Signs and symptoms
The syndrome gets its name from the characteristic cry of affected infants, which is similar to that of a meowing kitten, due to problems with the larynx and nervous system. About one third of children lose the cry by age of 2 years. Other symptoms of cri du chat syndrome may include:
- feeding problems because of difficulty in swallowing and sucking;
- mutism;
- low birth weight and poor growth;
- severe cognitive, speech and motor disabilities;
- behavioural problems such as hyperactivity, aggression, outbursts and repetitive movements;
- unusual facial features, which may change over time;
- excessive drooling;
- small head (microcephaly) and jaw (micrognathism);
- widely-spaced eyes (hypertelorism);
- skin tags in front of eyes.
Other common findings include hypotonia, a round face with full cheeks, epicanthal folds, down-slanting palpebral fissures (eyelids), strabismus, flat nasal bridge, down-turned mouth, low-set ears, short fingers, single palmar creases and cardiac defects (e.g., ventricular septal defect [VSD], atrial septal defect [ASD], patent ductus arteriosus [PDA], tetralogy of Fallot). Infertility is not associated with Cri du chat.
It has also been observed that people with the condition have difficulties communicating. While levels of proficiency can range from a few words to short sentences, it is often recommended by medical professionals for the child to undergo some sort of speech therapy/aid with the help of a professional.
Less frequently encountered findings include cleft lip and palate, preauricular tags and fistulas, thymic dysplasia, intestinal malrotation, megacolon, inguinal hernia, dislocated hips, cryptorchidism, hypospadias, rare renal malformations (e.g., horseshoe kidneys, renal ectopia or agenesis, hydronephrosis), clinodactyly of the fifth fingers, talipes equinovarus, pes planus, syndactyly of the second and third fingers and toes, oligosyndactyly and hyper extensible joints. The syndrome may also include various dermatoglyphics, including transverse flexion creases, distal axial triradius, increased whorls and arches on digits and a single palmar crease.
Late childhood and adolescence findings include significant intellectual disability, microcephaly, coarsening of facial features, prominent supraorbital ridges, deep-set eyes, hypoplastic nasal bridge, severe malocclusion and scoliosis.
Affected females reach puberty, develop secondary sex characteristics and menstruate at the usual time. The genital tract is usually normal in females, except for a report of a bicornuate uterus. In males, testes are often small, but spermatogenesis is thought to be normal.
Exceptionally, some with Cri du chat are very high-functioning and do not seem very different from developmentally typical individuals, with mostly the exception of mild learning difficulties, and do not have speech difficulties, although they may have milder facial features and a high-pitched voice due to their condition.
Genetics

Cri du chat syndrome is due to a partial deletion of the short arm of chromosome number 5, also called "5p monosomy" or "partial monosomy." Approximately 90% of cases result from a sporadic, or randomly occurring, de novo deletion. The remaining 10–15% are due to unequal segregation of a parental balanced translocation where the 5p monosomy is often accompanied by a trisomic portion of the genome. These individuals may have more severe disease than those with isolated monosomy of 5p. A recent study suggests this may not be the case where a trisomy of chromosome 4q is involved.[6]
Most cases involve total loss of the most distal 10–20% of the material on the short arm. Fewer than 10% of cases have other rare cytogenetic aberrations (e.g., interstitial deletions, mosaicisms, rings and de novo translocations). The deleted chromosome 5 is paternal in origin in about 80% of de novo cases. Loss of a small region in band 5p15.2 (cri du chat critical region) correlates with all the clinical features of the syndrome with the exception of the catlike cry, which maps to band 5p15.3 (catlike critical region). The results suggest that 2 noncontiguous critical regions contain genes involved in this condition's cause. Two genes in these regions, Semaphorine F (SEMA5A) and delta catenin (CTNND2), are potentially involved in cerebral development. The deletion of the telomerase reverse transcriptase (hTERT) gene localized in 5p15.33 may contribute to the phenotypic changes in cri du chat syndrome as well.
Diagnosis
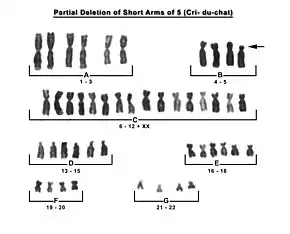
Diagnosis is based on the distinctive cry and accompanying physical problems. These common symptoms are quite easily observed in infants. Affected children are typically diagnosed by a doctor at birth. Genetic counseling and genetic testing may be offered to families with individuals who have cri du chat syndrome. Prenatally the deletion of the cri du chat related region in the p arm of chromosome 5 can be detected from amniotic fluid or chorionic villi samples with BACs-on-Beads technology. G-banded karyotype of a carrier is also useful.[7]
Treatment
There is not a specific way to treat the condition as the brain damage caused by this condition occurs in the early stages of embryo development. Intensive treatment is rarely needed in infants and they can be treated in neonatal pathology departments. Children may be treated by speech, physical and occupational therapists. If infants have difficulty in suction or swallowing, then physical therapy should begin in the first weeks of life. Heart abnormalities often require surgical correction and specialist attention.[8]
Prognosis
Once the child has survived the first few years of life, the prognosis is good and the mortality level is low. In a series of case reports, the mortality rate was about 10%, 75% of deaths occurring within 3 months of birth, and 90% within the 1st year.[8]
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 "Cri du chat syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 18 March 2021. Retrieved 25 March 2021.
- 1 2 3 4 5 6 "Cri du Chat Syndrome". NORD (National Organization for Rare Disorders). 2017. Archived from the original on 21 March 2021. Retrieved 25 March 2021.
- 1 2 Cerruti Mainardi, Paola (2006). "[No title found]". Orphanet Journal of Rare Diseases. 1 (1): 33. doi:10.1186/1750-1172-1-33.
- 1 2 3 4 5 6 7 "Learning About Cri du Chat". www.genome.gov. Archived from the original on 2015-12-10. Retrieved 2015-12-10.
- ↑ Lejeune J, Lafourcade J, Berger R, et al. (1963). "[3 Cases of partial deletion of the short arm of chromosome 5]". C. R. Acad. Sci. (in français). 257: 3098–102. PMID 14095841.
- ↑ Sheth, Frenny; Gohel, Naresh; Liehr, Thomas; Akinde, Olakanmi; Desai, Manisha; Adeteye, Olawaleye; Sheth, Jayesh (2012-01-01). "Gain of Chromosome 4qter and Loss of 5pter: An Unusual Case with Features of Cri du Chat Syndrome". Case Reports in Genetics. 2012: 153405. doi:10.1155/2012/153405. ISSN 2090-6544. PMC 3539376. PMID 23320207.
- ↑ "Cri-du-chat Syndrome". Medscape. 9 June 2017. Archived from the original on 25 August 2017. Retrieved 25 August 2017.
- 1 2 Cerruti Mainardi, Paola (2006-09-05). "Cri du Chat syndrome". Orphanet Journal of Rare Diseases. 1: 33. doi:10.1186/1750-1172-1-33. ISSN 1750-1172. PMC 1574300. PMID 16953888.
External links
Cri du chat at Curlie
| Classification | |
|---|---|
| External resources |
|