Endolymphatic sac tumor
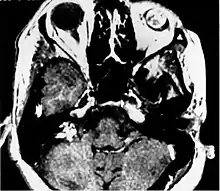
An endolymphatic sac tumor (ELST) is a very uncommon papillary epithelial neoplasm arising within the endolymphatic sac or endolymphatic duct. This tumor shows a very high association with von Hippel-Lindau syndrome (VHL).[1]
Classification
The ELST has been referred to as adenocarcinoma of endolymphatic sac, Heffner tumor,[1] papillary adenomatous tumor, aggressive papillary adenoma, invasive papillary cystadenoma, and papillary tumor of temporal bone. However, these names are not encouraged as they do not accurately classify the current understanding of the tumor.[2][3]
Signs and symptoms
Patients with ELST may present clinically with progressive or fluctuating, one sided sensorineural hearing loss which may mimick Meniere's disease due to the development of tumor associated endolymphatic hydrops.[4] Patients may also experience tinnitus, vertigo, and loss of vestibular function (ataxia). Alternatively, symptom onset may be sudden, due to intralabyrinthine hemorrhage.[4] Patients may also present with other symptoms related to von Hippel-Lindau syndrome in other anatomic sites, which will result in imaging evaluation of the head.[1][3][5]
Imaging findings
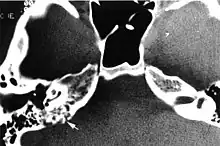
Imaging studies help to identify the tumor and the specific anatomic site of involvement. Magnetic resonance images show a hyperintensity (hypervascularity) of a heterogeneous mass by T1 weighted images. Computed tomography shows a multilocular, lytic destructive temporal bone mass, centered on the vestibular aqueduct (between internal auditory canal and sigmoid sinus).[1][6][7]
Pathogenesis
The von Hippel-Lindau tumor suppressor gene generally has a germline mutation. This suppressor gene is also called elongin binding protein and G7 protein. The VHL protein is involved in up-regulation of hypoxic response via the [[hypoxia inducible factor [HIF]-1 alpha]]. Mutations generally prevent the production of any functional VHL protein or result in a change of structure of VHL protein. This genetic disorder shows an autosomal dominant inheritance pattern, with about 20% of patients possessing a new mutation. There are usually several other tumors which are part of the syndrome, including tumors of the central nervous system, kidneys, pancreas, adrenal glands, epididymis, broad ligament, along with the endolymphatic sac. The vast majority of patients with an endolymphatic sac tumor have von Hippel-Lindau syndrome.[8][9]
Pathology findings
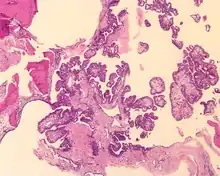
Tumors range from several millimeters up to 10 cm, with larger tumors more frequently seen in older patients. If the tumor is bilateral, it is almost always seen in a VHL patient. The tumor destroys the mastoid air spaces and extends into the middle ear and/or posterior cranial fossa.[1][3]
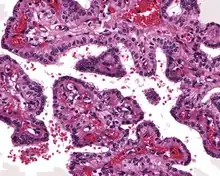
The microscopic appearance shows an unencapsulated, destructive growth, remodeling and invading bone. The tumor is arranged as simple, broad, non-complex papillary projections without large cystic spaces. The spaces are often fluid filled, have extravasated erythrocytes and/or inspissated material. The cells are cuboidal, usually single layered along the papillary structures, showing indistinct cell borders. The nuclei are round and hyperchromatic.[1][3][10][11]
Immunohistochemistry
The neoplastic cells are reactive with keratin, CK7, and Epithelial membrane antigen, but negative with TTF-1 and CK20.[10]
Cytogenetics
The germline mutations of VHL tumor suppressor gene will be found on 3p25-26 (short arm of chromosome 3), usually between base pair 10,158,318 to 10,168,761.[8][12]
Differential Diagnosis
The clinical and pathology differential are different. From a pathology perspective, an endolymphatic sac tumor needs to be separated from metastatic renal cell carcinoma, metastatic thyroid papillary carcinoma, middle ear adenoma, paraganglioma, choroid plexus papilloma, middle ear adenocarcinoma, and ceruminous adenoma.[1]
Management
Wide excision is the treatment of choice, although attempting to preserve hearing. Based on the anatomic site, it is difficult to completely remove, and so while there is a good prognosis, recurrences or persistence may be seen. There is no metastatic potential. Patients who succumb to the disease, usually do so because of other tumors within the von Hippel-Lindau complex rather than from this tumor.[1][13]
Epidemiology
This is a very rare tumor, since only about 1 in 35,000 to 40,000 people have VHL, of whom about 10% have endolymphatic sac tumors. Patients usually present in the 4th to 5th decades without an gender predilection. The tumor involves the endolymphatic sac, a portion of the intraosseous inner ear of the posterior petrous bone.[1][5]
References
- 1 2 3 4 5 6 7 8 9 Heffner DK (Dec 1989). "Low-grade adenocarcinoma of probable endolymphatic sac origin A clinicopathologic study of 20 cases". Cancer. 64 (11): 2292–302. doi:10.1002/1097-0142(19891201)64:11<2292::AID-CNCR2820641119>3.0.CO;2-#. PMID 2804921.
- ↑ Devaney KO, et al. (Dec 2003). "Endolymphatic sac tumor (low-grade papillary adenocarcinoma) of the temporal bone". Acta Otolaryngol. 123 (9): 1022–6. doi:10.1080/00016480310000494. PMID 14710902. S2CID 27359344.
- 1 2 3 4 Batsakis JG, et al. (Aug 1993). "Papillary neoplasms (Heffner's tumors) of the endolymphatic sac". Ann. Otol. Rhinol. Laryngol. 102 (8 pt 1): 648–51. doi:10.1177/000348949310200815. PMID 8352492. S2CID 24543165.
- 1 2 Butman, JA; Kim, HJ; Baggenstos, M; Ammerman, JM; Dambrosia, J; Patsalides, A; Patronas, NJ; Oldfield, EH; Lonser, RR (4 July 2007). "Mechanisms of morbid hearing loss associated with tumors of the endolymphatic sac in von Hippel-Lindau disease". JAMA. 298 (1): 41–8. doi:10.1001/jama.298.1.41. PMID 17609489.
- 1 2 Rodrigues S, et al. (Jul 2004). "Endolymphatic sac tumors: a review of the St. Vincent's hospital experience". Otol. Neurotol. 25 (4): 599–603. doi:10.1097/00129492-200407000-00030. PMID 15241241. S2CID 12656126.
- ↑ Connor SE, et al. (May 2008). "Imaging of the petrous apex: a pictorial review". Br. J. Radiol. 81 (965): 427–35. doi:10.1259/bjr/54160649. PMID 18208855.
- ↑ Leung RS, et al. (Jan–Feb 2008). "Imaging features of von Hippel-Lindau disease". Radiographics. 28 (1): 65–79. doi:10.1148/rg.281075052. PMID 18203931.
- 1 2 Glasker S, et al. (Dec 2005). "Effects of VHL deficiency on endolymphatic duct and sac". Cancer Res. 65 (23): 10847–53. doi:10.1158/0008-5472.CAN-05-1104. PMID 16322231.
- ↑ Richard S, et al. (Mar 2000). "Central nervous system hemangioblastomas, endolymphatic sac tumors, and von Hippel-Lindau disease". Neurosurg. Rev. 23 (1): 1–22. doi:10.1007/s101430050024. PMID 10809480. S2CID 41261476.
- 1 2 Horiguchi H, et al. (Jul 2001). "Endolymphatic sac tumor associated with a von Hippel-Lindau disease patient: an immunohistochemical study". Mod. Pathol. 14 (7): 727–32. doi:10.1038/modpathol.3880380. PMID 11455007.
- ↑ Kempermann G, et al. (Jul 1998). "Endolymphatic sac tumours". Histopathology. 33 (1): 2–10. doi:10.1046/j.1365-2559.1998.00460.x. PMID 9726042. S2CID 8791079.
- ↑ Richards FM, et al. (Jun 1998). "Molecular genetic analysis of von Hippel-Lindau disease". Journal of Internal Medicine. 243 (6): 527–33. doi:10.1046/j.1365-2796.1998.00334.x. PMID 9681854. S2CID 38520998.
- ↑ Megerian CA, et al. (Jun 2007). "Evaluation and management of endolymphatic sac and duct tumors". Otolaryngologic Clinics of North America. 40 (3): 463–78. doi:10.1016/j.otc.2007.03.002. PMID 17544692.
Further reading
Lester D. R. Thompson; Bruce M. Wenig (2011). Diagnostic Pathology: Head and Neck: Published by Amirsys. Hagerstown, MD: Lippincott Williams & Wilkins. pp. 7:64–67. ISBN 978-1-931884-61-7.