GM2 gangliosidoses
| GM2 gangliosidoses | |
|---|---|
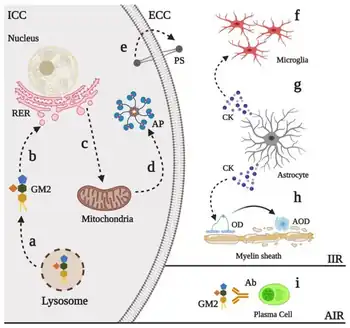 | |
| Physiopathological events in GM2 gangliosidoses | |
The GM2 gangliosidoses are a group of three related genetic disorders that result from a deficiency of the enzyme beta-hexosaminidase. This enzyme catalyzes the biodegradation of fatty acid derivatives known as gangliosides.[1] The diseases are better known by their individual names: Tay–Sachs disease, AB variant, and Sandhoff disease.
Beta-hexosaminidase is a vital hydrolytic enzyme, found in the lysosomes, that breaks down lipids. When beta-hexosaminidase is no longer functioning properly, the lipids accumulate in the nervous tissue of the brain and cause problems. Gangliosides are made and biodegraded rapidly in early life as the brain develops. Except in some rare, late-onset forms, the GM2 gangliosidoses are fatal.[1]
All three disorders are rare in the general population. Tay–Sachs disease has become famous as a public health model because an enzyme assay test for TSD was discovered and developed in the late 1960s and early 1970s, providing one of the first "mass screening" tools in medical genetics. It became a research and public health model for understanding and preventing all autosomal genetic disorders.[2][3]
Tay–Sachs disease, AB variant, and Sandhoff disease might easily have been defined together as a single disease, because the three disorders are associated with failure of the same metabolic pathway and have the same outcome. Classification and naming for many genetic disorders reflects history, because most diseases were first observed and classified based on biochemistry and pathophysiology before genetic diagnosis was available. However, the three GM2 gangliosidoses were discovered and named separately. Each represents a distinct molecular point of failure in a subunit that is required for activation of the enzyme.
Tay–Sachs disease
Tay–Sachs disease is a rare autosomal recessive genetic disorder that causes a progressive deterioration of nerve cells and of mental and physical abilities that begins around six months of age and usually results in death by the age of four. It is the most common of the GM2 gangliosidoses. The disease occurs when harmful quantities of cell membrane gangliosides accumulate in the brain's nerve cells, eventually leading to the premature death of the cells.[4]
Sandhoff disease
Sandhoff disease is a rare, autosomal recessive metabolic disorder that causes progressive destruction of nerve cells in the brain and spinal cord. The disease results from mutations on chromosome 5 in the HEXB gene, critical for the lysosomal enzymes beta-N-acetylhexosaminidase A and B. Sandhoff disease is clinically indistinguishable from Tay–Sachs disease. The most common form, infantile Sandhoff disease, is usually fatal by early childhood.[5]
AB variant
GM2-gangliosidosis, AB variant is a rare, autosomal recessive metabolic disorder that causes progressive destruction of nerve cells in the brain and spinal cord. Mutations in the GM2A gene cause AB variant. The GM2A gene provides instructions for making a protein called the GM2 activator. This protein is a cofactor that is required for the normal function of beta-hexosaminidase A. The disease is usually fatal by early childhood.[6]
Diagnosis
In terms of the evaluation we find that in the case of Sandhoff disease , is tested via beta-hexosaminidase A and beta-hexosaminidase B enzyme activity; those with the disease have reduced or no activity.[7]
Treatment
There are no authorized therapies for the treatment of the GM2 Gangliosidosis (Tay-Sachs and Sandhoff disease).[8] The current standard of care for GM2 Gangliosidosis disease is limited to supportive care and aimed at providing adequate nutrition and hydration.[9]
This supportive care may substantially improve the quality of life of people affected by GM2. The therapeutic team may include specialists in neurology, pulmonology, gastroenterology, psychiatrist, orthopaedics, nutrition, physical therapy and occupational therapy.
N-Acetyl-Leucine
N-Acetyl-Leucine is an orally administered, modified amino acid that is being developed as a novel treatment for multiple rare and common neurological disorders by IntraBio Inc (Oxford, United Kingdom).[10]
N-Acetyl-Leucine has been granted multiple orphan drug designations from the U.S. Food & Drug Administration (FDA)[11] and the European Medicines Agency (EMA)[12] and the European Medicines Agency (EMA) for the treatment of a various genetic diseases, including GM2 Gangliosidosis (Tay-Sachs and Sandhoff). The US FDA has granted IntraBio a Rare Pediatric Disease Designation for N-Acetyl-Leucine for the treatment of GM2 Gangliosidosis.[13]
Compassionate use studies in both Tay-Sachs and Sandhoff patients have demonstrated the positive clinical effects of treatment with N-Acetyl-Leucine for GM2 Gangliosidosis[14] These studies further demonstrated that the treatment is well tolerated, with a good safety profile.
A multinational clinical trial investigating N-Acetyl-L-Leucine for the treatment of GM2 Gangliosidosis (Tay-Sachs and Sandhoff) began in 2019[15] Recruitment is ongoing.
IntraBio is also conducting parallel clinical trials with N-Acetyl-L-Leucine for the treatment of Niemann-Pick disease type C[16] and Ataxia-Telangiectasia.[17] Future opportunities to develop N-Acetyl-Leucine include Lewy body dementia,[18] amyotrophic lateral sclerosis, restless leg syndrome, multiple sclerosis, and migraine.[19]
See also
- GM2 (ganglioside)
References
- 1 2 Mahuran DJ (1999-10-08). "Biochemical consequences of mutations causing the GM2 gangliosidoses". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1455 (2–3): 105–138. doi:10.1016/S0925-4439(99)00074-5. PMID 10571007.
- ↑ O'Brien JS, Okada S, Chen A, Fillerup DL (1970). "Tay–Sachs disease. Detection of heterozygotes and homozygotes by serum hexaminidase assay". New England Journal of Medicine. 283 (1): 15–20. doi:10.1056/NEJM197007022830104. PMID 4986776.
- ↑ Kaback MM (2001). "Screening and prevention in Tay–Sachs disease: origins, update, and impact". Advances in Genetics. 44: 253–65. doi:10.1016/S0065-2660(01)44084-3. ISBN 978-0-12-017644-1. PMID 11596988.
- ↑ "Biomarker for Gangliosidosis: BioGM1/BioGM2 (BioGM1/GM2) (BioGM1/BioGM2)". ClinicalTrials.gov Archive. NIH US National Library of Medicine. Archived from the original on 2 May 2021. Retrieved 2 May 2021.
- ↑ "Sandhoff Disease". Medline Plus. NIH U.S. National Library of Medicine. Archived from the original on 8 April 2010. Retrieved 2 May 2021.
- ↑ Bley, Annette E. (Nov 2011). "Natural History of Infantile GM2 Gangliosidosis". Pediatrics. 128 (5): e1233–e1241. doi:10.1542/peds.2011-0078. PMC 3208966. PMID 22025593.
- ↑ "Sandhoff Disease - Symptoms, Causes, Treatment | NORD". rarediseases.org. Archived from the original on 18 March 2023. Retrieved 18 October 2023.
- ↑ Patterson, Marc C. (2013-01-01), Dulac, Olivier; Lassonde, Maryse; Sarnat, Harvey B. (eds.), "Chapter 174 - Gangliosidoses", Handbook of Clinical Neurology, Pediatric Neurology Part III, Elsevier, 113: 1707–1708, doi:10.1016/B978-0-444-59565-2.00039-3, ISBN 9780444595652, PMID 23622392, archived from the original on 2019-08-01, retrieved 2019-08-01
- ↑ Kaback, Michael M.; Desnick, Robert J. (1993). "HEXA Disorders". In Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.). Hexosaminidase A Deficiency. GeneReviews®. University of Washington, Seattle. PMID 20301397. Archived from the original on 2014-01-16. Retrieved 2019-08-01.
- ↑ "IntraBio". Archived from the original on 2019-08-01. Retrieved 2019-08-01.
- ↑ "Search Orphan Drug Designations and Approvals". www.accessdata.fda.gov. Archived from the original on 2021-04-27. Retrieved 2019-08-01.
- ↑ Anonymous (2018-09-17). "EU/3/17/1949". European Medicines Agency. Archived from the original on 2019-08-01. Retrieved 2019-08-01.
- ↑ "IntraBio". October 2018. Archived from the original on 2019-08-01. Retrieved 2019-08-01.
- ↑ "Tanganil in patients with tay-sachs and sandhoff". Hand in Hand gegen Tay-Sachs und Sandhoff | in Deutschland e.V. (in Deutsch). Retrieved 2019-08-01.
{{cite web}}: CS1 maint: url-status (link) - ↑ "N-Acetyl-L-Leucine for GM2 Gangliosdisosis (Tay-Sachs and Sandhoff Disease)". clinicaltrials.gov. Archived from the original on 2021-08-07. Retrieved 2019-08-01.
- ↑ "N-Acetyl-L-Leucine for Niemann-Pick Disease, Type C (NPC)". clinicaltrials.gov. Archived from the original on 2019-08-01. Retrieved 2019-08-01.
- ↑ "N-Acetyl-L-Leucine for Ataxia-Telangiectasia". clinicaltrials.gov. Archived from the original on 2019-08-01. Retrieved 2019-08-01.
- ↑ "IntraBio". Archived from the original on 2019-08-01. Retrieved 2019-08-01.
- ↑ Strupp, Michael; Bayer, Otmar; Feil, Katharina; Straube, Andreas (2019-02-01). "Prophylactic treatment of migraine with and without aura with acetyl-dl-leucine: a case series". Journal of Neurology. 266 (2): 525–529. doi:10.1007/s00415-018-9155-6. ISSN 1432-1459. PMID 30547273. S2CID 56148131.
External links
- GeneReview/NIH/UW entry on Hexosaminidase A Deficiency Archived 2014-01-16 at the Wayback Machine
| Classification | |
|---|---|
| External resources |
|